Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
#12023-04-05 05:54:24
- sanjukta
- Member
- Registered: 2023-04-05
- Posts: 1
TrESP analysis in Multifwn, convergence error
Hello Multiwfn Team,
I was curious to obtain the intermolecular Coulomb interaction energy from the TrESP charges using cubegen utility. I have used the keywords mentioned in the manual to generate the gaussian checkpoint files. The method and keywords were as follows #p td=(nstates=10) wb97xd/6-31+g(d,p) nosymm IOp(9/40=4) scf=xqc.
The calculation was done for a pi-stacked dimer system, the problem came, when we had fixed the the bottom monomer unit and moved the second unit along the short-axis, while the magnitude of the coupling decays gradually with longer distances, there was an abrupt sign change of the interacation energy along the short-axis shift. I am attaching the image of the Coulomb interaction energy (eV) wrt to shift in short-axis here. Can you please help me adressing this error.
Image link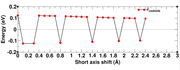
Link to the drive with files
https://drive.google.com/drive/folders/ … sp=sharing
Last edited by sanjukta (2023-04-05 05:55:51)
Offline
#22023-04-05 12:20:49

Re: TrESP analysis in Multifwn, convergence error
Currently it is inconvenient for me to check your files. What I can suggest is checking the sign of TrESP charges, perhaps the sign shows somewhat arbitrariness. If after flipping the sign of the TrESP charges for the abrupt changed points the curve will be smooth, please just use flipped result.
Offline