Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
#12018-04-01 12:01:36
- Halaou
- Member
- Registered: 2018-03-22
- Posts: 2
The overlap between HONTO and LUNTO
Hi everybody,
I've performed a TDDFT calculation and I've successfully calculated the integral overlap between HOMO and LUMO. Now I'm looking forward to calculating the overlap between HONTO and LUNTO at singlet (S1) and triplet (T1) states. Could you guid me please to performe it?
Thank you.
Offline
#22018-04-01 14:29:01

Re: The overlap between HONTO and LUNTO
Hi,
You can firstly use Multiwfn to generate NTOs (see example in Section 4.18.4), then use main function 0 to find the index corresponding to the so-called HONTO and LUNTO (the energies printed on console window now correspond to NTO eigenvalues), and then use subfunction 10 of main function 200 to evaluate overlap integral between the two orbitals.
Best regards,
Tian
Offline
#32018-04-02 10:28:08
- Halaou
- Member
- Registered: 2018-03-22
- Posts: 2
Re: The overlap between HONTO and LUNTO
Hello,
Thanks for the reply. Concerning the IOp, should I use the same used in the example "IOp(9/40=4)" ? I'm using a Gaussian16 rev A03.
Offline
#42018-04-02 13:04:12
Re: The overlap between HONTO and LUNTO
Hello,
Thanks for the reply. Concerning the IOp, should I use the same used in the example "IOp(9/40=4)" ? I'm using a Gaussian16 rev A03.
Yes, if you intend to use Multiwfn to generate NTOs, IOp(9/40=4) is needed, so that enough configuration state coefficients could be printed in Gaussian output file.
Offline
#52020-11-07 21:34:30
- Rania14
- Member
- Registered: 2020-11-01
- Posts: 7
Re: The overlap between HONTO and LUNTO
Hello,
1- Please what does it mean IOp(9/40=4) exactly, what I will have the new in my output.
2- Should I use IOp(9/40=4) in every Job Type or only in the optimization?
Thank you
Offline
#62020-11-07 22:46:24
Re: The overlap between HONTO and LUNTO
Hello,
1- Please what does it mean IOp(9/40=4) exactly, what I will have the new in my output.
2- Should I use IOp(9/40=4) in every Job Type or only in the optimization?
Thank you
This point has been mentioned in the beginning of Section 3.21:
Gaussian users: Output file (.out or .log) of CIS, TDHF, TDDFT and TDA-DFT tasks can be used. Both single point and optimization tasks are supported; for the latter case, Multiwfn analyzes electronic excitation at the final geometry. Since by default Gaussian only outputs the configuration coefficients whose absoluate value is larger than 0.1, In order to achieve acceptable accuracy, you must add IOp(9/40=4) keyword in the route section so that all configuration coefficients whose magnitude larger than 0.0001 will be printed (If the calculation in Multiwfn is found to be too expensive, using IOp(9/40=3) instead is also generally acceptable).
If you need to analyze excited states using the methods that rely on configuration coefficients (e.g. hole-electron analysis, NTO, IFCT, transition density matrix...), this keyword should be used.
Offline
#72020-11-07 22:52:58
- Rania14
- Member
- Registered: 2020-11-01
- Posts: 7
Re: The overlap between HONTO and LUNTO
Ah okey, thank you, so I will use IOp(9/40=4) in every steps (optimization, energy, frequency...)
I'm wondering if Multiwfn can calculate the ground state oxidation potential, I really need it to calculate the driving force (ΔGinject).
Offline
#82020-11-08 10:20:39
Re: The overlap between HONTO and LUNTO
Ground state oxidation potential should be calculated by quantum chemistry code. See e.g.https://dx.doi.org/10.1021/acs.jpca.0c05052
Offline
#92021-10-13 03:13:12
- Usman Ali
- Member
- Registered: 2021-10-13
- Posts: 13
Re: The overlap between HONTO and LUNTO
Hi,
You can firstly use Multiwfn to generate NTOs (see example in Section 4.18.4), then use main function 0 to find the index corresponding to the so-called HONTO and LUNTO (the energies printed on console window now correspond to NTO eigenvalues), and then use subfunction 10 of main function 200 to evaluate overlap integral between the two orbitals.
Best regards,
Tian


Hi
After reading your reply to the this post, I have calculated the overlap of hole and electron between the HONTO and LUNTO but the answer value which I got from the Multiwfn is very different from the result like 0.00000000... something like that. I want to know should we need to multiply or accomodate this value with any factor?
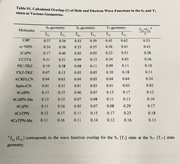
Secondly, I want to ask you that I want to calculate the overlap for different states with different geometries. So, I need to calculate the NTOs via guassian calculations for every state like S1 and T1 from different geometries like S0, S1, T1, T2, T3 etc. I have also attached the figures with this question. I want to perform the calculations given in the Table S1 of the picture 2 in this post. I should be respected for your guidance and response.
Thanks
Last edited by Usman Ali (2021-10-14 01:56:09)
Offline
#102021-10-13 12:43:04
Re: The overlap between HONTO and LUNTO
1 NTOs are orthogonal with each other, so it is obvious that overlap integral between two different NTOs must be an extremely small value (exactly zero, in principle). To reproduce the data in your photo, you should use hole-electron analysis module of Multiwfn and take Sm index:
 (screenshot of Section 3.21.1 of multiwfn manual)
(screenshot of Section 3.21.1 of multiwfn manual)
See Section 4.18.1 of Multiwfn manual for example on how to use hole-electron analysis module.
2 You do not need to calculate NTO at all. In fact NTO doesn't work in many cases, it is very often to find there is no NTO pair dominates the excitation of interest. In contrast, the hole-electron analysis in Multiwfn is universal and rigorous, the overlap between hole and electron measured by Sm index can faithfully describe the overlap extent in any situation. See Section 3.21.1 of Multiwfn manual for theory of hole-electron analysis.
You can respectively optimize the S0, S1, T1 geometries, and at each geometry, perform hole-electron analysis to derive Sm index for S0-S1 and S0-T1 excitations, respectively.
Offline
#112021-10-14 02:06:54
- Usman Ali
- Member
- Registered: 2021-10-13
- Posts: 13
Re: The overlap between HONTO and LUNTO
1 NTOs are orthogonal with each other, so it is obvious that overlap integral between two different NTOs must be an extremely small value (exactly zero, in principle). To reproduce the data in your photo, you should use hole-electron analysis module of Multiwfn and take Sm index:
https://i.postimg.cc/68FzFCXH/Clipboard01.png(screenshot of Section 3.21.1 of multiwfn manual)
See Section 4.18.1 of Multiwfn manual for example on how to use hole-electron analysis module.
2 You do not need to calculate NTO at all. In fact NTO doesn't work in many cases, it is very often to find there is no NTO pair dominates the excitation of interest. In contrast, the hole-electron analysis in Multiwfn is universal and rigorous, the overlap between hole and electron measured by Sm index can faithfully describe the overlap extent in any situation. See Section 3.21.1 of Multiwfn manual for theory of hole-electron analysis.
You can respectively optimize the S0, S1, T1 geometries, and at each geometry, perform hole-electron analysis to derive Sm index for S0-S1 and S0-T1 excitations, respectively.
Thank you very much for your answer. I can not clearly understand your mean in the last line as you said "just optimize the different geometries and at each geometry to drive the Sm index". You mean from optimized geomtries, I need to calculate their TDDFT calculations for excitations and then calculate their electron-hole overlap extent?
Secondly, you mentioned that NTO are not suitable for calculating the overlap extent so if I used the simple molecular orbitals like HOMO and LUMO for calculating the overlap extent, then even the results are very small as close to zero.
I think there is a confusion for my understanding, so could you please explain it in more details.
Thank you very much for this forum to help us.
Last edited by Usman Ali (2021-10-19 02:03:31)
Offline
#122021-10-14 04:27:00
Re: The overlap between HONTO and LUNTO
Thank you very much for your answer. I can not clearly understand your mean in the last line as you said "just optimize the different geometries and at each geometry to drive the Sm index". You mean from optimized geomtries, I need to calculate their TDDFT calculations for excitations and then calculate their electron-hole overlap extent?
Secondly, you mentioned that NTO are not suitable for calculating the overlap extent so if I used the simple molecular orbitals like HOMO and LUMO for calculating the overlap extent, then even the results are very small as close to zero.
I think there is a confusion for my understanding, so could you please explain it in mnore details.
Thank you very much for this forum to help us.
1 Yes
2 There are several points you need to know:
(1) Hole-electron analysis in Multiwfn takes all orbital transitions into account and thus rigorous in all cases. So, the excitation to be characterized is not necessarily fully dominated by single pair of MO or NTO transition.
(2) There are different ways to measure overlap between hole and electron (given by hole-electron analysis in Multiwfn), or between two orbitals. For the former, you can use Sr or Sm index directly printed by hole-electron analysis module; for the latter, you can use Multiwfn to calculate overlap integral of their norm or their squares, this point is mentioned in Section 3.100.11 of Multiwfn manual.

(3) Overlap integral between two orbitals is completely different to overlap integral betweennormof two orbitals. The latter is able to quantify overlap extent of spatial distributions of two orbitals, while the former is exactly zero if the orbitals are orthogonal to each other (e.g. MO, NTO) and thus useless to quantify orbital overlap. In #9, you used Multiwfn to calculate the former.
Offline
#132021-10-19 02:15:44
- Usman Ali
- Member
- Registered: 2021-10-13
- Posts: 13
Re: The overlap between HONTO and LUNTO

Thank you soo much. After your detail guidance, I have found the Sm index for overlap. I would like to know that Sm value is in a.u units, So it would require to chnage this value into eV units or its should be use as such. Also, tell me which units are better for its explanation as it is not mentioned in mannual. The picture I send you before for reproductions of such results not explains any unit. Kindly guide me in this regard.
Thanks
Offline
#142021-10-19 14:53:55
Re: The overlap between HONTO and LUNTO
Thank you soo much. After your detail guidance, I have found the Sm index for overlap. I would like to know that Sm value is in a.u units, So it would require to chnage this value into eV units or its should be use as such. Also, tell me which units are better for its explanation as it is not mentioned in mannual. The picture I send you before for reproductions of such results not explains any unit. Kindly guide me in this regard.
Thanks
Sm is not a quantity of energy, so obviously it cannot be represented using eV. You should always use a.u. as unit.
As you can see from your screenshot, the unit has already been clearly given, namely a.u. (atomic unit)
Offline
#152021-10-21 02:39:52
- Usman Ali
- Member
- Registered: 2021-10-13
- Posts: 13
Re: The overlap between HONTO and LUNTO
Thank you very much for your help. I have completed this analysis.
Offline