Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
#12021-05-07 03:56:54
- shenaya
- Member
- Registered: 2019-10-16
- Posts: 34
Calculating emission spectra
Dear Tian,
I need to plot emission spectra of Ru transition metal complexes.
This is my input for optimization.
Is this correct?
I need both fluorescence and phosphorescence.
Can I take the output of this calculation to obtain the emission spectra using Multiwfn
%mem=4GB
%nprocshared=4
%chk=Neutral.chk
#p td=(50-50,nstates=10,root=1) b3lyp/genecp scrf=(solvent=methanol)
guess=save geom=connectivity opt
Neutral Fluoride Complex from the X-ray crystal structure
0 1
Ru 0.49152900 -1.00287600 0.09326700
Cl 0.66506000 -1.18796400 -2.33847900
Cl 0.96242300 -1.06595000 2.50569800
P -1.14911300 0.54149000 0.02331800
F 2.76034900 -1.90860800 0.00631700
N 1.77532400 0.64178100 -0.02843200
O -2.22603700 -2.70087200 0.96397000
S -0.87762300 -2.87042900 0.29049200
C 1.27960300 1.90403300 -0.05083200
C -0.13813900 2.08851300 -0.01943800
C -0.64790200 3.36905700 0.00649500
H -1.72172700 3.52421800 0.05440300
C 0.20504300 4.50762600 -0.02719900
H -0.23475300 5.49948700 -0.01111100
C 1.56632600 4.35252600 -0.08013500
H 2.22538600 5.21520600 -0.10762500
C 2.14873400 3.04896800 -0.08989900
C 3.53085900 2.83652700 -0.12465600
H 4.20665800 3.68669100 -0.15556800
C 4.05139700 1.53821100 -0.11568800
C 5.45120700 1.25536100 -0.15050600
H 6.14675600 2.08773100 -0.19077900
C 5.90357800 -0.03875900 -0.13249900
H 6.96691600 -0.25133600 -0.15894000
C 4.98721500 -1.12783200 -0.07836500
H 5.33654100 -2.15449100 -0.06390000
C 3.65038400 -0.86564600 -0.04418000
C 3.11234700 0.44907000 -0.06204300
C -1.19758300 -3.75212800 -1.26942600
H -1.79914200 -4.63222900 -1.02933400
H -0.25052300 -4.02752200 -1.73610500
H -1.74514100 -3.07082800 -1.92117300
C -0.00292000 -4.20020300 1.17802700
H 0.23376000 -3.81407100 2.17029700
H 0.91641400 -4.44979400 0.64186000
H -0.66776000 -5.06540600 1.23910000
C -2.18596100 0.55229000 -1.50150700
C -3.18113500 -0.43327200 -1.62938500
H -3.34446200 -1.14905100 -0.82796800
C -3.96636500 -0.49594700 -2.78073900
H -4.73480500 -1.25924300 -2.86603300
C -3.76744600 0.42012100 -3.81820500
H -4.38023800 0.37087500 -4.71387100
C -2.78018900 1.39921300 -3.69735000
H -2.62036000 2.11581000 -4.49811900
C -1.99099500 1.46594500 -2.54554700
H -1.22701800 2.23231700 -2.46900500
C -2.38962000 0.87519600 1.35444700
C -3.60606500 1.51256500 1.05151600
H -3.85995700 1.75348300 0.02521400
C -4.50566300 1.84038400 2.06810900
H -5.44108100 2.33239300 1.81668500
C -4.20643400 1.53429500 3.39706000
H -4.90946000 1.78576800 4.18632200
C -3.00006700 0.90154900 3.70558200
H -2.76052400 0.65709200 4.73687400
C -2.09536100 0.57430500 2.69378500
H -1.16252800 0.08069500 2.94054800
1 4 1.0 8 1.0
2
3
4 10 1.0 37 1.0 48 1.0
5 27 1.0
6 9 1.5 28 1.5
7 8 1.5
8 29 1.0 33 1.0
9 10 1.5 17 1.5
10 11 2.0
11 12 1.0 13 1.5
12
13 14 1.0 15 2.0
14
15 16 1.0 17 1.5
16
17 18 1.5
18 19 1.0 20 1.5
19
20 21 1.5 28 1.5
21 22 1.0 23 2.0
22
23 24 1.0 25 1.5
24
25 26 1.0 27 2.0
26
27 28 1.5
28
29 30 1.0 31 1.0 32 1.0
30
31
32
33 34 1.0 35 1.0 36 1.0
34
35
36
37 38 1.5 46 1.5
38 39 1.0 40 1.5
39
40 41 1.0 42 1.5
41
42 43 1.0 44 1.5
43
44 45 1.0 46 1.5
45
46 47 1.0
47
48 49 1.5 57 1.5
49 50 1.0 51 1.5
50
51 52 1.0 53 1.5
52
53 54 1.0 55 1.5
54
55 56 1.0 57 1.5
56
57 58 1.0
58
C H P N F S O 0
6-31G(d)
****
Cl
6-31+G(d)
****
Ru 0
SDD
****
Ru 0
SDD
Thanks and regards,
Gayani
Offline
#22021-05-07 07:38:24

Re: Calculating emission spectra
Use following keywords for optimizing S1
#p td b3lyp/genecp scrf=(solvent=methanol) geom=connectivity opt
Use following keywords for optimizing T1
#p td(triplet) b3lyp/genecp scrf=(solvent=methanol) geom=connectivity opt
Note that S1 and T1 must be optimized separately, 50-50 cannot be used in this context. guess=save is meaningless. It is unnecessary to set nstates as large as 10, because this will notably increase computational cost while the S1 / T1 will not be detectably affected.
Offline
#32021-05-07 08:29:57
- shenaya
- Member
- Registered: 2019-10-16
- Posts: 34
Re: Calculating emission spectra
Thank you, Tian. Will do it. Generally how many nstates should be used for this type of transition metal complexes.
Offline
#42021-05-07 17:23:15
Re: Calculating emission spectra
Usually, nstates should be at least root+2, where the root is the state of interest. If you optimize S1, then root=1 is of interest, therefore nstates should be >=3. By default, the nstates is just 3, therefore you do not need to alter the default nstates setting. Increasing nstates is not harmful on the result, but higher computational cost is needed.
Offline
#52021-05-11 01:32:04
- shenaya
- Member
- Registered: 2019-10-16
- Posts: 34
Re: Calculating emission spectra
Thank you very much !!!
Offline
#62021-05-14 02:46:41
- shenaya
- Member
- Registered: 2019-10-16
- Posts: 34
Re: Calculating emission spectra
Hi Tian,
I have successfully plot fluorescence spectra by following the instructions stated by you (//www.umsyar.com/wfnbbs/viewtopic.php?id=413).
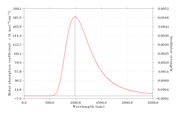
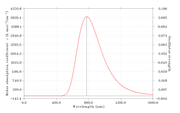
1. My question is can we plot phosphorescence spectra using exactly same steps?
11 // Plotting spectra
3 // UV-Vis
20 // Modify oscillator strengths
2-3 // Choose S2 and S3 states
0 // Set oscillator strengths to zero (because emission is from S1 state)
0 // Plot spectrum
Becouse phosphorescence is T1---->S0 transition.
Another thing is when people mean emission spectra which one in transition metal complexes.
fluorescence, phosphorescence, or both?
Thank you
Offline
#72021-05-14 04:38:39
Re: Calculating emission spectra
No, you can't.
Gaussian is able to calculate emission energy of phosphorescence spectrum, but it is unable to yield oscillator strength of this kind of spectrum (in other words, the printed oscillator strength is always exactly zero), since non-vanishing oscillator strength of T1->S0 emission comes from spin-orbit coupling effect, which cannot be considered by Gaussian at TDDFT level. Usually Dalton program is suggested to compute oscillator strength between T1 and S0.
I cannot understand your second question.
Offline
#82021-05-25 10:41:40
- shenaya
- Member
- Registered: 2019-10-16
- Posts: 34
Re: Calculating emission spectra
Dear Tian,
This is a part of the output file for fluorescence emission calculation.
Input : #p opt td=(singlets,root=1) b3lyp/genecp scrf=(solvent=methanol) geom=
connectivity
I have calculated emission spectra. When do orbital contribution calculation I have to consider transition from 145 ---->147 ?
or I understood this wrongly?
Excitation energies and oscillator strengths:
Excited State 1: Singlet-A 1.2577 eV 985.84 nm f=0.0047
145 ->147 0.68259
This state for optimization and/or second-order correction.
Total Energy, E(TD-HF/TD-KS) = -3027.47515828
Copying the excited state density for this state as the 1-particle RhoCI density.
Excited State 2: Singlet-A 1.7817 eV 695.89 nm f=0.0001
144 ->147 -0.67710
Excited State 3: Singlet-A 1.9683 eV 629.89 nm f=0.0027
145 ->146 0.69194
SavETr: write IOETrn= 770 NScale= 10 NData= 16 NLR=1 NState= 3 LETran= 64.
The selected state is a singlet
Offline
#102021-05-27 00:59:30
- shenaya
- Member
- Registered: 2019-10-16
- Posts: 34
Re: Calculating emission spectra
Thank you.
Offline