Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
#12019-03-05 10:25:55
- julieborah66
- Member
- Registered: 2018-09-17
- Posts: 32
Low quality grid points
Sir,
For calculating the ESP of an octamer (that contains 104 atoms) the total number of grid points used for calculating the ESP of an atom is 10,000 grid points. Is this a reasonable value of grid points? Since, increasing it would cause computational longer time and also to be noted that my system is bigger.
Offline
#22019-03-05 18:00:03

Re: Low quality grid points
Dear julieborah66,
The ESP can be studied in many different ways, I don't know which way did you want to adapt. If it is difficult to describe, please show me all commands that you intended to input in Multiwfn.
Tian
Offline
#32019-03-05 18:51:48
- julieborah66
- Member
- Registered: 2018-09-17
- Posts: 32
Re: Low quality grid points
Thanks Sir for your prompt reply.
Here are my details, I want to calculate the RMSE of the .chg file with Quantum ESP using MK fitting mode
There is an option of selecting the MK layers in the menu, I want to select all the points lying in between 1.66 of the vdw radius to 2.20 of the vdw radius of a particular atom. I am giving the layers option as 1.66,1.67, 1.68...till 2.20, so 55 layers. Then it yields a statement that 11020 points are being used. I am attaching some pics of the screen where commands are mentioned.
So, I would like to know from you if I am doing it the correct way?
If this method is wrong is there any way to do this of getting RMSE of a specific atom within a particular range of vdw radius specified by user, using any fitting method in Multiwfn?
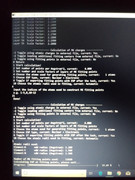
Last edited by julieborah66 (2019-03-05 19:17:27)
Offline
#42019-03-05 19:09:03
- julieborah66
- Member
- Registered: 2018-09-17
- Posts: 32
Re: Low quality grid points
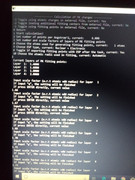
Offline
#52019-03-05 23:54:16
Re: Low quality grid points
From the screenshot the settings should be correct.
Since the status of option 6 has been switched to "Yes", after the calculation, you can select if exporting the fitting points. You can export them as .pqr file, then load the file into VMD, from the graphical window if you find the distribution of the fitting points meets your expectation, that means your setting must be meaningful.
Offline
#62019-03-06 05:04:04
- julieborah66
- Member
- Registered: 2018-09-17
- Posts: 32
Re: Low quality grid points
Okay Sir, Thank you for helping me out. I will look at the distribution of points in VMD.
Offline