Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
#12024-07-01 11:32:10
- e119340
- Member
- Registered: 2023-10-06
- Posts: 8
Calculation of charges with ghost atoms
Dear friends,
I need to calculate the CHELPG charges for the O2 molecule in a 3-site model (2 O atoms and a ghost atom at the center of mass).
Would it be possible to do this in Multiwfn? I.e., calculate the charges considering a ghost atom?
If anyone is familiar with the psi4 software, which output from the psi4 software should I use as input to the Multiwfn software to calculate CHELPG charges?
Thanks in advance.
Offline
#22024-07-01 16:30:13
- e119340
- Member
- Registered: 2023-10-06
- Posts: 8
Re: Calculation of charges with ghost atoms
Complementing: my idea is to obtain partial charges because I am developing a force field to be used in molecular dynamics simulations.
Offline
#32024-07-03 05:31:31

Re: Calculation of charges with ghost atoms
The "ghost atom" you mentioned is referred to "additional fitting center" in the context of ESP fitting in Multiwfn. You can find option "-2 Toggle loading additional fitting centers from external file" in CHELPG interface (subfunction 12 of main function 7), by which you can fit point charges that not located at nuclei. Please check Section "4.7.7.6 Example 6: RESP charge calculation with additional fitting centers" of Multiwfn manual (this section used RESP interface, while the consideration of additional fitting centers is exactly the same as CHELPG interface)
You can use .fch file produced by PSI4 as input file of Multiwfn for this purpose. Please check beginning part of Chapter 4 of Multiwfn manual on how to generate it by PSI4.
Offline
#42024-07-03 18:53:56
- e119340
- Member
- Registered: 2023-10-06
- Posts: 8
Re: Calculation of charges with ghost atoms
Thank you very much for your kindness, Tian Lu.
I will investigate the points you mentioned in the manual.
Multiwfn has a lot of features, I'm impressed!
Best,
Offline
#52024-07-04 03:31:59
- e119340
- Member
- Registered: 2023-10-06
- Posts: 8
Re: Calculation of charges with ghost atoms
I have one final question.
I calculated the charges for O2 considering an additional fitting center. For the CHELPG method, the O charges differed slightly, while for the RESP the charges of the O values were the same.
Is there any way that I can force the O charges for the CHELPG method to be equal? Or is this difference already expected in this method?
Results for CHELPG: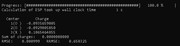
Results for RESP: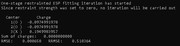
Offline
#62024-07-04 09:53:02
Re: Calculation of charges with ghost atoms
By default the RESP module employs the same fitting points as MK method, the distribution of the fitting points in this case has the same symmetry as O2 molecular symmetry, therefore the resulting atomic charges are exactly the same. However, the distribution of CHELPG fitting points doesn't satisfy this condition, so there is an expected marginal difference between the two oxygen charges, you can simply take their average, or use MK/CHELPG interface to derive the atomic charges.
Offline
#72024-08-12 14:02:28
- e119340
- Member
- Registered: 2023-10-06
- Posts: 8
Re: Calculation of charges with ghost atoms
Hello, I have an additional question about this topic.
For a 4-site model of H2O (like in the figure), in which the charges are placed on the H atoms and at point M. Is there any way to not consider the O atom and only at point M in the charges calculation?
I know how to place an additional fitting center at point M but I don't know how to disregard atom point O. How could I do this in Multiwfn?
Thanks in advance.
Best,
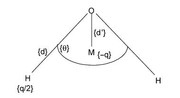
Offline
#82024-08-12 19:31:44
Re: Calculation of charges with ghost atoms
In the RESP module of Multiwfn you can set charge constraint(s) using option "6 Set charge constraint in fitting", I think if you constraint the charge of the oxygen to zero, you can realize your aim.
Offline