Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 Re: Multiwfn and wavefunction analysis » calculating the cavity diameter » 2024-10-11 07:37:48
I am using both multiwfn 3.7 and multiwfn 3.8 dev. software but none of them takes the "cav" input. Is there any updated version that I can perhaps download?
Thank in advance
Regards
Louis-Charl
#2 Multiwfn and wavefunction analysis » calculating the cavity diameter » 2024-10-10 16:40:10
- Louischarlc0
- Replies: 3
Dear Prof. Lu
Hope you are good. I'm trying to calculate the cavity parameter but when I enter "cav" as stated in the manual then it dies on me, in spite of uploading a Pdb file. What can I do to fix this?
Thanks in advance
Regards
Louis-Charl
#3 Re: Multiwfn and wavefunction analysis » Charge Transfer Spectrum » 2024-02-24 14:25:08
It seems to work now. Thank you so much. I really appreciate your assistance
Regards
Louis-Charl
#4 Re: Multiwfn and wavefunction analysis » Charge Transfer Spectrum » 2024-02-24 13:10:30
and I boot up multiwfn by double clicking on the application software icon
Regards
Louis-Charl
#5 Re: Multiwfn and wavefunction analysis » Charge Transfer Spectrum » 2024-02-24 13:08:45
I'm using Windows 11 Pro
#6 Re: Multiwfn and wavefunction analysis » Charge Transfer Spectrum » 2024-02-23 20:03:32
At first, the fchk file that I use as input file wasn't in the subfolder as the multiwfn directory and therefore generated the "CT_multiple" in the same folder as this fchk folder. Then I decided to copy it into the multiwfn directory but it did not work. Then I decided to delete the "CT_multiple" folder in the other directory so that I can use the one in the multiwfn directory but it did not work either. Do you think I should move the fchk file into the multiwfn directory and repeat the process of generating the"CT_multiple" folder?
Thanks in advance
Regards
Louis-Charl
#7 Multiwfn and wavefunction analysis » Charge Transfer Spectrum » 2024-02-23 17:43:36
- Louischarlc0
- Replies: 7
Dear Prof Lu
I seems to get this error message "Error: Cannot find CT_multiple\total_spectrum.txt" when I upload the input file "CT_multiple\CT_multiple.txt" after I selected option 3 for the UV-Vis even though it generated 5 input files and produced this "CT_multiple\total_spectrum.txt UV-Vis" in the first line of CT_multiple.txt file. Why is this the case?
Thanks in advance
Regards
Louis-Charl
#8 Re: Multiwfn and wavefunction analysis » multiplicity » 2022-12-17 10:31:26
Dear Prof Lu
This is the sintex that I used to optimized them. They optimized successfully on Gaussian.
#p opt freq m062x/genecp geom=connectivity int=(grid=ultrafine,acc2e=12) scf=(xqc,tight,maxcycles=1000) scrf=(solvent=methanol)
Thanks in advance
Regards
Louis-Charl
#9 Multiwfn and wavefunction analysis » multiplicity » 2022-12-16 16:55:46
- Louischarlc0
- Replies: 3
Dear Prof Lu
I want to create N.wfn; N-1.wfn and N+1.wfn input files for CDFT analysis but it looks like copper and iodine containing compounds are problematic. How can I fix this?
Thanks in advance
Regards
Louis-Charl
#10 Re: Multiwfn and wavefunction analysis » molden » 2022-03-02 15:54:11
I want to study the intermolecular interactions between neighboring molecules within a cluster.
#11 Re: Multiwfn and wavefunction analysis » molden » 2022-03-02 09:59:06
The link below is what I got from VMD when I select an atom within the supercell cluster by following the steps in section 4.2 of the tutorial.
#12 Re: Multiwfn and wavefunction analysis » molden » 2022-03-01 16:05:41
I'm struggling to upload the file. Maybe because its a notepad file.
Regards
Louis-Charl
#13 Multiwfn and wavefunction analysis » molden » 2022-03-01 12:03:49
- Louischarlc0
- Replies: 7
Dear Prof Lu
I followed the steps in section 4.1 of the tutorial and was able to generate a .molden file. As I was unable to open this molden file directly with multiwfn, I decided to edited it via notepad and enter the [cell] information that are supplied in this tutorial. However, I was still unable to open this file via multiwfn and found out that previous molden files e.g. A.molden, AT.molden etc. that I was able to open via multiwfn were also affected. Is this code only applicable for the H2O molecule that is described in the tutorial or can it be expanded to larger organic systems?
Thanks in advance
Regards
Louis-Charl
#14 Re: Multiwfn and wavefunction analysis » IGM » 2022-02-26 15:34:11
I think I've managed to sort it out now thanks.
Regards
Louis-Charl
#15 Re: Multiwfn and wavefunction analysis » IGM » 2022-02-26 15:22:01
cp2k. One of the steps in the tutorial says toggle exporting . molden file but it does not produce anything.
Regards
Louis-Charl
#16 Re: Multiwfn and wavefunction analysis » IGM » 2022-02-26 09:15:42
Dear Prof Lu
I'm struggling to produce the .molden file in section 4.1 of the tutorial. I've also read through section 2.9 of the manual. Is there any alternative on how you can produce the .molden file?
Thanks in advance
Regards
Louis-Charl
#17 Re: Multiwfn and wavefunction analysis » IGM » 2022-02-24 15:05:58
Thank you for your response. Are the steps on for sections 4.-4.3 in the IGMH tutorial applicable to large organic molecules also?
Thanks in advance
Regards
Louis-Charl
#18 Re: Multiwfn and wavefunction analysis » IGM » 2022-02-23 16:47:37
This is the image that I obtained after I uploaded the pdb file of a porphyrin structure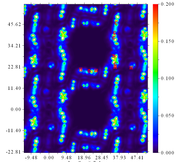
#19 Multiwfn and wavefunction analysis » IGM » 2022-02-23 11:00:29
- Louischarlc0
- Replies: 9
Dear Prof Lu
I'm struggling to obtain a well defined image as on page 768 for my compounds, although I've used different colour scales. What should I do to get a well-defined image?
Thanks in advance
Regards
Louis-Charl
#20 Re: Multiwfn and wavefunction analysis » CIF files » 2022-02-22 12:38:59
When I enter option 25 then it gives me the same options. Why is this the case?
Thanks in advance
Regards
Louis-Charl
#21 Re: Multiwfn and wavefunction analysis » CIF files » 2022-02-22 09:14:21
I followed the steps in section 4.12.6 yesterday, but after I input option 19 to construct the supercell, then it request me for replicas in direction. How many replicas do I need to input?
Thanks in advance
Regards
Louis-Charl
#22 Multiwfn and wavefunction analysis » CIF files » 2022-02-21 14:10:02
- Louischarlc0
- Replies: 5
Dear Prof Lu
If I use a CIF file as an input file will it be possible to obtain info about intermolecular strengths between monomers using multiwfn analysis?
Thanks in advance
Regards
Louis-Charl
#23 Multiwfn and wavefunction analysis » coordinating atoms » 2022-02-21 11:59:11
- Louischarlc0
- Replies: 1
Dear Prof Lu
When I perform the NOCV analysis then I receive this message
Error: Atom 1 of this fragment has evidently different coordinate to the corresponding atom ( 1 ) of the whole system!
X,Y,Z of atom 1 in this fragment: 0.07219 -2.10597 0.08904 Angstrom
X,Y,Z of atom 1 in whole system: 15.38680 7.32820 7.74390 Angstrom
How can I circumvent it?
Thanks in advance
Regards
Louis-Charl
#24 Multiwfn and wavefunction analysis » Orca » 2022-02-19 19:13:00
- Louischarlc0
- Replies: 1
Dear Prof Lu
To perform the ETS-NOCV I see you need Orca so that you can convert the gbw files to Molden files as input. I also notice in the multiwfn application software there is no option 23 to perform the ETS-NOCV. I tried to re-install the multiwfn package but unfortunately it wasn't formatted. As there are several versions of Orca software, which version can I install and how can I re-install a formatted multiwfn software with the updated multiwfn application software?
Thanks in advance
Regards
Louis-Charl
#25 Re: Multiwfn and wavefunction analysis » intermolecular orbital overlap » 2022-02-19 11:31:20
My apologies. I see you've updated the manual and found it page 816
Thank you
Regards
Louis-Charl
#26 Re: Multiwfn and wavefunction analysis » intermolecular orbital overlap » 2022-02-19 11:09:50
Thank you for your response. However, I do not see section 4.23.4 in the latest version of the multiwfn manual. Are you sure it is not section 4.20.4 as it contains the info that you are describing.
Regards
Louis-Charl
#27 Multiwfn and wavefunction analysis » intermolecular orbital overlap » 2022-02-18 11:30:14
- Louischarlc0
- Replies: 3
Dear Prof Lu
I've calculated the intermolecular orbital overlap for my dimer compound. However, I'm not sure how to interpret the result as the manual does not explain it on page 774. For both HOMO-HOMO and LUMO-LUMO overlaps I got negative results. As I'm under the impression that orbital overlap is related to the strength of interaction between monomers, what does it mean to get negative or positive result and what does the value of the result say about the magnitude of the interaction?
Thanks in advance
Regards
Louis-Charl
#28 Re: Multiwfn and wavefunction analysis » GNUplot command prompt » 2022-02-11 19:10:16
Hi there
What command prompt can I use to generate a high quality image for fukui function or dual descriptor when using VMD?
Thanks in advance
Regards
Louis-Charl
#29 Re: Multiwfn and wavefunction analysis » GNUplot command prompt » 2021-11-06 10:44:21
set terminal postscript landscape enhanced color 'Helvetica' 20
set encoding iso_8859_1
set output 'RDGscatter.ps'
set key
set ylabel 'RDG (a.u)' font "Helvetica, 20"
set xlabel 'sign({/Symbol l}_2){/Symbol r} (a.u.)' font "Helvetica, 20"
set pm3d map
set palette defined (-0.035 "blue",-0.0075 "green", 0.02 "red")
set format y "%.2f"
set format x "%.2f"
set format cb "%.3f"
set border lw 2
set xtic -0.05,0.01,0.05 nomirror rotate font "Helvetica"
set ytic 0.0,0.2,2.0 nomirror font "Helvetica"
set cbtic -0.035,0.005,0.02 nomirror font "Helvetica"
set xrange [-0.05:0.05]
set yrange [0.0:2.0]
set cbrange [-0.035:0.02]
plot 'output.txt' u 4:5:4 with points pointtype 31 pointsize 0.3 palette t ''
Above is the information from the notepad file
Regards
Louis-Charl
#30 Re: Multiwfn and wavefunction analysis » GNUplot command prompt » 2021-11-06 08:21:20
I managed to enter the cmd environment now and the command prompt seemed to work but it gives me a notepad file. Should I edit it to get the PS format of the 2D RDGscatter?
Thanks in advance
Regards
Louis-Charl