Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » NCI Isosurface question » 2024-12-08 16:03:53
I understand sir. In my case, i just got the "func1.cub" and "func2.cub files off multiwfn and opened them on vmd, using "source rdgfill.vmd". So i am using this color scheme of the script.
Thank you for your answer!
#2 Multiwfn and wavefunction analysis » NCI Isosurface question » 2024-12-06 18:04:31
- brunosamp
- Replies: 2
When i get an NCI isosurface with vdw interactions, i sometimes get these yellow-brownish color in my nci. I am not sure if this indicates and attractive vdW interaction, or a repulsive interaction. Could you clarify this to me?
Here is the image:
I am sending a URL with the image because i am getting an error with the "Postimage" website.
#3 Re: Multiwfn and wavefunction analysis » Question about comparasion in electron-hole analysis » 2024-03-10 16:00:19
Mr. tian lu, just one last question. Epoxy groups are known to induce non-radiative recombinations. Electron-hole analysis can show this type of behavior?
#4 Re: Multiwfn and wavefunction analysis » Question about comparasion in electron-hole analysis » 2024-03-05 13:59:59
I understand now, thank you!
#5 Multiwfn and wavefunction analysis » Question about comparasion in electron-hole analysis » 2024-03-04 17:36:58
- brunosamp
- Replies: 4
I have 4 coronene structures. One is only the coronene, and the other 3 are functionalized with: Only an OH groups (Cor-OH); Only an epoxy group (Cor-ep), and only a -COOH group (Cor-cooh).
I want to do a electron-hole analysis to characterize the td-dft absorption bands i obtained (If it's n - > pi*, pi -> pi*).
However, i'd also like to compare how the functional group affects the fluorescence of this structure.
To do so, i wanted to compare the charge density difference isosurfaces, to show the localization of electron and hole, and also the Dindex and transition dipole moment.
My question is: In the Cor-OH groups, the highest oscillator strength is in the S9 excitation, in the Cor-Ep is in the S12 and the Cor-COOH is the S11 excitation. I compared them, and all of them are LE excitations. Can i compare the transition dipole moments and the Dindex to try to compare which structure would have a greater emission in the fluorescence spectra?
Or do i have to pick the same excitation (Ex: The S1 excitation of all structures) to properly compare them? The S1 excitation i obtained for them are not observed experimentally, which is why i'm not using it.
#6 Re: Multiwfn and wavefunction analysis » Help with NCI analysis with integration » 2024-01-27 15:47:47
I Will do this sir, thank you very much
#7 Re: Multiwfn and wavefunction analysis » Help with NCI analysis with integration » 2024-01-24 18:34:22
Dear tian lu, i made a mistake while typing.
For the basal hydroxyl, i got a hydrogen bond interaction and a van der walls interaction as well.
NCI Isosurface: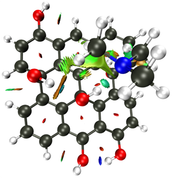
Closest domain to the NCI Isosurface i got, which described very well the NCI isosurface.
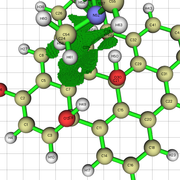
While for the edge hydroxyl, i got this nci isosurface (VdW interaction and the hydrogen bond with the edge hydroxyl):
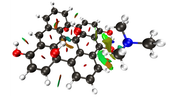
And this domain was the closest one i got:
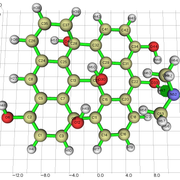
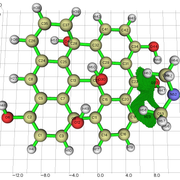
But in the nci, the hydrogen bond and vdw interaction are not separeted, like above. So, since the interaction with the edge hydroxyl is the strongest, i understand the integral value will not be well comparable with the integral value for the basal hydroxyl i showed above, i believe.
#8 Multiwfn and wavefunction analysis » Help with NCI analysis with integration » 2024-01-24 18:11:31
- brunosamp
- Replies: 4
I performed a nci analysis for a system of graphene oxide with a ammonia molecule interacting with the edge and basal hydroxyl.
In the NCI analysis, i observed that the interaction with the edge hydroxyl is described by a hydrogen bond region and weak van der walls interactions with the carbons near the hydroxyl. The same goes for the basal hydroxyl.
When i perform the integration for this structure, i got a list of domains. In the edge, one of the domains showed only the hydrogen bond interaction, and not the van der walls interaction. On the basal hydroxyl, i got a domain with the van der walls interaction and the hydrogen bond.
How can i get the exact same isosurface i got in the NCI analysis, when i do the integration?
Just to add context: the bond strength with the edge hydroxyl is higher than with the basal hydroxyl. But, with these domains i am getting, the integral of the interaction with the basal hydroxyl is higher than with the edge hydroxyl, which i understand it should be the opposite.
I used the default criterion for defining the domain (<0.5).
#9 Re: Multiwfn and wavefunction analysis » Question about electron-hole recombination heat map » 2023-08-31 16:24:11
Thank you sir, i was able to do it!
Just one more question, if i may. I have 5 graphene based structures, with slight structural differences (one has a hydroxyl group, another a carboxyl group, etc.). Each one of them has a different absorption range, which means that each structure has a different excited state with higher oscillator strength.
Can i compare all these structures using different excited states, or i'd need to use the same excited state, always?
#10 Multiwfn and wavefunction analysis » Question about electron-hole recombination heat map » 2023-08-29 16:52:06
- brunosamp
- Replies: 3
I want to do a electron-hole recombination heat map for a graphene structure. But i still don't understand the proper calculation to obtain it.
Based on the example provided by the manual, i need to do a TD-DFT calculation asking for a number of states:
Example: m062x/6-31+G(d,p) TD(nstates=n) IOP(9/40=4) and the ground state coordinates of my optimized molecule
The formchk of this calculation will be enough to obtain the heat map or i need to do another calculation?
Second question: Would the diffuse function of my method bring any sort of problems to obtain my heat map?
#11 Re: Multiwfn and wavefunction analysis » Charge density difference analysis for graphene » 2023-06-20 16:40:06
Sir, now i understand it. Thank you very much!
#12 Re: Multiwfn and wavefunction analysis » Charge density difference analysis for graphene » 2023-06-14 17:05:58
Tian lu, thank you for your response! One question: for the second method, shouldn't i measure the charge in the carbon, in which the OH group is attached, without the OH group?
For example: without the OH group, the charge of the carbon is -0.02. But after attaching the OH group, this same carbon has 0.065 charge (i used the method you mention in "2" for both structures). So, wouldn't the charge transfer be 0.085, from the carbon to the OH group?
#13 Multiwfn and wavefunction analysis » Charge density difference analysis for graphene » 2023-06-12 19:30:20
- brunosamp
- Replies: 4
I am studying the PDOS (Partial Density of States) of a graphene oxide structure, with only hydroxyl groups.
To study the PDOS, it's useful to evaluate the charge transfer from the carbon to the OH functional group.
I understood how to perform the charge density difference map in a excitation process. But in my case, i just want to evaluate how the electron density is distributed towards that OH group.
1) Can i obtain the isosurface of charge density difference, in my case, using the method in 4.18.3 section.
2) Can i measure the amount of charge transfer (Q) from the carbon to the OH group using Multiwfn? I couldn't find something about this in the manual.
#14 Re: Multiwfn and wavefunction analysis » Fluorescence or absorption output for electron-hole for analysis? » 2023-05-01 01:00:35
Yes, i chose the first excited state. I was using the fchk and output file from a absorption calculation, which i don't think it was correct for the analysis, right? I am now optimizing the excited geometry to do this analysis properly.
Just one more question, if i have a anti-kasha result, should i just choose another excited state?
#15 Multiwfn and wavefunction analysis » Fluorescence or absorption output for electron-hole for analysis? » 2023-04-30 16:41:57
- brunosamp
- Replies: 3
I am trying to analyse the electron-hole population for a pure graphene quantum dot (no functional group) and a graphene quantum dot with hydroxyls. I used M062X/6-31+G(d,p), and the hirshfeld method to calculate the electron-hole overlaps. To achieve this i used the output of the absorption calculation, which i used the following input, for both structures
#p td=(nstates=100, singlet) m062x/6-31+G(d,p) gfinput pop=FULL IOP(9/40=4) density=current
The result of the overlaps between electron and hole was higher for the pure graphene. And the transition dipole moments were:
Transition dipole moment in X/Y/Z: -0.001971 -2.531006 -0.000003 a.u. (Pure graphene)
Transition dipole moment in X/Y/Z: 0.090665 0.026479 0.361728 a.u (Graphene with OH group)
But the experimentation shows that functionalized graphene has a higher fluorescence intensity. I have two questions:
1) I should've used the output of the fluorescence for this analysis?
2) The negative transition dipole moment of graphene means that the graphene with the OH graphene would have higher fluorescence intensity?
#16 Re: Multiwfn and wavefunction analysis » Multiwfn can analyze the orbitals involved in each UV/transition? » 2023-04-30 01:53:27
Thank you very much for your help, Mr. Tian Lu!
#17 Multiwfn and wavefunction analysis » Multiwfn can analyze the orbitals involved in each UV/transition? » 2023-04-27 14:56:39
- brunosamp
- Replies: 2
I am trying to analyze the orbitals that are involved in the absorption transitions of my TD-DFT analysis.
I used m062x/6-31G(d,p), under TD-DFT, to obtain the absorption spectrum of my molecule.
But i want to analyise, for example, if the transition in a certain wavelength is of the type "n->pi* or "pi->pi*".
Can the multiwfn do this type of analysis?
#18 Re: Multiwfn and wavefunction analysis » How to properly define fragments for PDOS graph? » 2023-04-22 16:23:00
Now it is very clear to me.
Thank you very much, sir!
#19 Re: Multiwfn and wavefunction analysis » How to properly define fragments for PDOS graph? » 2023-04-21 22:03:10
Sir, you answer helped me greatly. Thank you very much!
Just one more question, if you could answer me. When i make a DOS/PDOS graph to three similar systems, i get graphs with the fermi energy in different ranges of temperature. I see many articles that present this fermi energy in zero.
The correct procedure is to normalize all my graphs to present a fermi energy=0?
#20 Multiwfn and wavefunction analysis » How to properly define fragments for PDOS graph? » 2023-04-21 18:10:23
- brunosamp
- Replies: 4
I am doing a PDOS graph for a graphene quantum dot (GQDs) molecule. This GQDs has hydroxyl, epoxy and carboxyl groups.
I don't understand how to define the fragments in my case:
1) For epoxy groups, should i include the carbons attached to the oxygens (C-O-C) or only the oxygens?
2) For the hydroxyl group, should i include the hydrogen of the O-H, and the carbon attached to this group, or only the oxygen?
3) For the carboxyl, should i include the whole COOH group, or only the oxygens?
This is important because it affects the PDOS graph a lot.
Thank you in advance.
Pages: 1