Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Multiwfn and wavefunction analysis » TrESP analysis in Multifwn, convergence error » 2023-04-05 05:54:24
- sanjukta
- Replies: 1
Hello Multiwfn Team,
I was curious to obtain the intermolecular Coulomb interaction energy from the TrESP charges using cubegen utility. I have used the keywords mentioned in the manual to generate the gaussian checkpoint files. The method and keywords were as follows #p td=(nstates=10) wb97xd/6-31+g(d,p) nosymm IOp(9/40=4) scf=xqc.
The calculation was done for a pi-stacked dimer system, the problem came, when we had fixed the the bottom monomer unit and moved the second unit along the short-axis, while the magnitude of the coupling decays gradually with longer distances, there was an abrupt sign change of the interacation energy along the short-axis shift. I am attaching the image of the Coulomb interaction energy (eV) wrt to shift in short-axis here. Can you please help me adressing this error.
Image link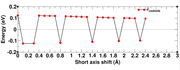
Link to the drive with files
https://drive.google.com/drive/folders/ … sp=sharing
Pages: 1