Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Quantum Chemistry » NMR Spectra Simulation Gaussian 16 » 2023-07-18 08:36:00
okok
do you know why Gaussian 16 doesn't show me the spin multiplicity in simulated spectra ?
i didn't find anything in literature.
Thanks
#2 Re: Quantum Chemistry » NMR Spectra Simulation Gaussian 16 » 2023-07-17 12:33:36
How can i define it the spin multiplicity in Gaussian Input file?
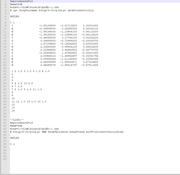
This is the input file: i have checked with Gaussian 16 User Manual and i think it's correct.
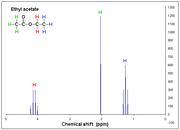
This is the experimental spectrum of ethyl acetate and the one simulated with Gaussian, for which I cannot see the spin multiplicity is shown above.
Thanks a lot
Alessio M.
#3 Re: Quantum Chemistry » NMR Spectra Simulation Gaussian 16 » 2023-07-17 10:56:35
Thank you so in your opinion with MULTIWFN i wil be able to see SPIN MULTIPLICITY?
thanks a lot
Alessio Macorano
#4 Re: Quantum Chemistry » NMR Spectra Simulation Gaussian 16 » 2023-07-17 07:19:07
Dear Prof Tian Lu,
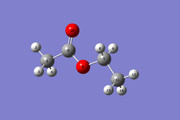
yes i have used the net charge and multiplicity 0 1. here i attached two picture: one of the molecular structure and the NMR simulated spectra with Gaussian 16. level of theory: B3LYP/ 6-31+G(2d,p) 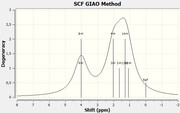
Thank you so much
Alessio M.
#5 Quantum Chemistry » NMR Spectra Simulation Gaussian 16 » 2023-07-14 10:34:43
- AlessioM
- Replies: 9
Dear Prof Tian lu.
i'm a PhD student in medicinal chemistry at University of Milan, IT.
As a collaboration with other organic chemistry i'm trying to simulate NMR spectra with Gaussian 16, and i'm reaching you out because i'm facing with a problem on this topic.
I'd like to Ask you if you ever have any problem to see spin multiplicity in NMR spectra from Gaussian16.
Because for a simple molecule like Ethyl Acetate Gaussian doesn't show me the multiplicity spin. (The ppm are rescaleted by TMS reference and they are correct respect to sperimental spectra).
I mean it doesn't show me Doublet, triplet and so on... I have used many DFT functionals and different basis set.
I'm very courious about It. If you want i can share with you .gjf, log, chk files.
Thanks for your suggestion
Alessio Macorano
#6 Re: Multiwfn and wavefunction analysis » Topological index » 2023-06-08 09:55:32
Dear Prof Tian Lu,
as far as i know this kier-hall index, also know as connectivity index, is a type of a molecular descriptor that is calculated based on the molecular graph of a chemical compound.Molecular graph is a representation in which the atoms are vertices and bonds are the edges.
I have found in literature that similar index to topological already cited are: Quantum Similarity Related Indices (QSRI).
As reported here: "Quantum similarity topological indices definition, analysis and comparison with classical molecular topological indices"
paper by Emili Besalú (Universitat de Girona) Ramon Carbó-Dorca (Universitat de Girona) january 1995, sorry but i can't find the DOI.
Maybe this can clarify my previous question.
Thanks a lot
Alessio
#7 Multiwfn and wavefunction analysis » Topological index » 2023-06-08 07:26:51
- AlessioM
- Replies: 3
Dear Tian Lu,
I'd like to ask you if it is possible to get a topological index similar to the Kier-Hall topological index using MULTIWFN?
If not, could you suggest me a way ? or other descriptors relatively close to the topological index?
I'd like to have such an index because I'd like to use it as molecular descriptors, I have .log and .fchk files of full optimised geometry.
Thanks a lot
My Best
Alessio
#8 Quantum Chemistry » Energy from gaussian output files » 2023-03-15 13:55:29
- AlessioM
- Replies: 1
Dear Prof Tian Lu,
I have a question about the energy value obtained from the Gaussian output file.
I've done opt + freq calculation on dichloromethane at B3LYP 6-31+G* as an example and I get an E(B3LYP)=-959.700528 HARTREE/PARTICLE, I know that this energy is related to complete dissociation reaction of the molecule.
If i use a 1 Hartree = 627.5 kcal/mol i get a huge number to be an energy value, is this correct?
Or is there another way to obtain an energy value in kcal/mol more reliable by common sense?
Thanks a lot !
Alessio Macorano
#9 Multiwfn and wavefunction analysis » Fukui function in "frozen core approximation" » 2022-12-15 14:38:26
- AlessioM
- Replies: 1
Dear Prof. Tian Lu
I have calculated condensed fukui function as in the example 4.22 (Phenol molecule) with finite difference method approach.
Is there any way to calculate fukui function with "frozen core approximation" with multiwfn ?
(i.e homo density for electrophilic attacks and lumo density for nucleophilic attacks calculation?)
If not possible, where can i extract those values?
I appreciate a lot !
Best regards !
Alessio
#10 Multiwfn and wavefunction analysis » generation of .wfn files for fukui function calculation » 2022-10-13 08:32:24
- AlessioM
- Replies: 1
Dear Multiwfn Users
Regarding to calculation of fukui function following phenol examples in 4.22 section:
is there any way to speed up production of N.wfn, N-1.wfn and N+1.wfn from formatted checkpoint file (.fhck)?
Do you know which features can accellerate this process? maybe use faster pc or with more cores, memory?
I appreciate a lot!
best regards
Alessio
#11 Multiwfn and wavefunction analysis » Superdelocalizability index » 2022-10-11 16:45:18
- AlessioM
- Replies: 1
Good morning to everyone
First of all thanks to the developers of Multiwfn works very well.
I have one question about superdelocalizability index:
As reported in the manual those index can be calculate for closed shell system, for my case i need that also for open shell system (because in our study we would like to compare them with semiempirical calculation) someone can help me?
thank you
bye!
Pages: 1