Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
Pages:1
#1Re:Multiwfn and wavefunction analysis»help»2023-11-06 05:23:54
Thanks for confirming my query.
krjt
#2Re:Multiwfn and wavefunction analysis»help»2023-11-03 09:46:19
Thank you very much.
Sorry to bother you.
How to get the molden.input file for ground state? Is it from ground state optimization file, ex., opt.gbw?
krjt
#3Re:Multiwfn and wavefunction analysis»help»2023-11-02 16:40:09
This is a fairly good tutorial for density difference map between excited state and ground state from the outputs of Gaussian calculations.
How to generate wfn files required for this task from ORCA calculations?
Your help will be much appreciated.
krjt
Dear mest2309,
The way of generating density difference map between excited state and ground state in fact has been substantially illustrated in Section 4.18.3 of the manual.
The easiest way to do this is generating .wfn file for S0 and S1, respectively, and then use custom operation of main function 5 to plot the isosurface map.
First, optimize your molecule at S0 state to generate the S0 geometry, and then use below input file to generate .wfn file for S0:
# B3LYP/6-311G** out=wfn
test
0 1
[Optimized S0 geometry]C:\S0.wfn
Then use this file to generate .wfn for S1
# B3LYP/6-311G** TD density out=wfn
test
0 1
[Optimized S0 geometry]C:\S1.wfn
Next, boot up Multiwfn and input following commands
C:\S1.wfn // Load excited state wavefunction file
5 // Generate grid data
0 // Set custom operation
1
-,C:\S0.wfn // Property of ground state wavefunction will be substracted from that of excited state wavefunction
1 // The property to be studied is electron density
2 // Medium quality grid (if your system is large, select 3 to use high quality grid to get better result)
-1 // Visualize isosurfaceThen you will see density difference isosurface map corresponding to rho(S1)-rho(S0) at S0 geometry, the map exhibits the charge redistribution during vertical excitation. You can adjust "isovalue" parameter in the GUI window to make the graph looks better.
Best regards,
Tian
#4Multiwfn and wavefunction analysis»excited state similarity in HONTO (sH) and LUNTO (sL)»2023-10-18 08:15:01
- krjt
- Replies: 1
-
Can we use multiwfn to calculate excited state similarity in HONTO (sH) and LUNTO (sL) as described in J. Mater. Chem. C, 2019, 7, 9523.
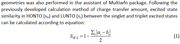
#5Re:Multiwfn and wavefunction analysis»There is a problem on batch analysis of multiple output files»2022-03-16 18:10:16
yes Prof. Lu. I was using Multiwfn 3.7. Now I installed 3.8 dev. It is working fine in that.
Thanks.
#6Multiwfn and wavefunction analysis»There is a problem on batch analysis of multiple output files»2022-03-15 16:47:11
- krjt
- Replies: 2
-
I faced a peculiar problem with MultiWfn.
I had to analyze the output of two tddft calculations.
I have used the following sequence of inputs:
outfile1
18
15
n
0
r
outfile2
18
15
n
0
q
The results printed on the screen for the second analysis (outfile2) was same as that obtained for outfile1.
It appears that the program is not resetting the parameters while loading a new output file using the command 'r'.
I had faced the same problem on using batch method with the above sequence of input.
Both the tddft output files were generated as suggested.
Justin
#7Re:Multiwfn and wavefunction analysis»Is there a method in Multiwfn to calculate φS Descriptor»2022-02-18 06:12:40
I understand. Thank you very much.
Justin
#8Re:Multiwfn and wavefunction analysis»Is there a method in Multiwfn to calculate φS Descriptor»2022-02-18 05:41:12
Thank you Prof. Lu Tian for your suggestion. I have been using recently Multiwfn for the calculation of Sr and Sm and related indices.
It appears that you have not included the PhiS index calculation in Multiwfn. It is fine.
Thanks again.
Justin
#9Multiwfn and wavefunction analysis»Is there a method in Multiwfn to calculate φS Descriptor»2022-02-17 13:02:57
- krjt
- Replies: 4
-
Is it possible to calculate patial overlap between detachment and attachment densities (φS Descriptor) as described in J. Chem. Theory Comput. 2014, 10, 3896−3905.
Thanks for the support.
Justin
Pages:1