Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
Pages:1
#1Re:Multiwfn and wavefunction analysis»Missing bond between some atoms in VMD»2022-10-07 14:36:16
Dear Prof. Lu,
I found an easier way to make missing bonds between some atoms. One someone run "source aim.vmd" and then "source igm_inter.vmd" and saw some missing bonds it is easier to remove the "bond paths" between atoms from graphics menu and then make the missing bond and then again activate the "bond paths".
Regards,
Reza
#2Re:Multiwfn and wavefunction analysis»Missing bond between some atoms in VMD»2022-08-29 07:18:33
Dear Prof. Lu,
many thanks.
Reza
#3Multiwfn and wavefunction analysis»Missing bond between some atoms in VMD»2022-08-28 03:51:48
- rkia
- Replies: 5
-
Dear Prof. Lu,
I obtained the output of AIM and IGM routines and put them in VMD for visualization by source aim.vmd and source igm_inter.vmd for a metal complex. Some of the bonds are missing in VMD and I tried to make the bond between those atoms via "Mouse" and "addd and remove bond" in VMD but it doesn't work. Could you please guide me.
Regards,
Reza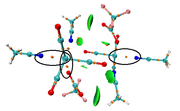
#4Re:Multiwfn and wavefunction analysis»Strange ESP map»2021-12-03 20:29:44
Dear Prof. Lu,
Yes, I did as you mentioned. I generated the surface.
Thanks
#5Re:Multiwfn and wavefunction analysis»Strange ESP map»2021-12-03 19:03:17
Dear Prof. Lu,
Many thanks for your kind help. I adjusted the color scale in the ESPiso.vmd script in the vmd folder but again why all of the B factors in the surfaceanalysis.pdb are positive.
I didn't do the analysis by reading the .wfn file and I followed your video manual by clicking on ESPpt.bat, ESPiso.bat, and ESPext.bat, respectively to obtain the necessary files to read in vmd.
Many thanks,
Reza
#6Re:Multiwfn and wavefunction analysis»Strange ESP map»2021-12-03 11:19:00
Many thanks.
#7Re:Multiwfn and wavefunction analysis»Strange ESP map»2021-12-03 11:14:37
Dear Prof. Lu,
I obtained the correct ESP with MultiWFN version 2021-Sep 13 but I didn't give the same results with MultiWFN 2021, Nov. 28. I also checked the ESPiso, ESPpt, ESPext.bat.... and related.vmd files in both MultiWFN and VMD folders to be sure I copied based on the new version but again the result was a totally red colored surface.
I attach the first ESP I obtained a few weeks ago and will send you the .fchk file.
This is the pic of the ESP I obtained before:-
Regards,
Reza
#8Multiwfn and wavefunction analysis»Strange ESP map»2021-12-03 06:59:47
- rkia
- Replies: 8
-
Dear Prof. Lu,
I calculated the ESP by reading 1.fchk file of Gaussian via ESPpt.vmd, ESPiso.vmd, and ESPext.vmd routines in MultiWFN but the map is in single colour and all B factors in the Surfaceanalysis.pdb file are positive.
##################
REMARK Unit of B-factor field (i.e. ESP) is kcal/mol
HETATM 1 C MOL A 1 -4.995 -0.702 1.818 1.00 64.59 C
HETATM 2 C MOL A 1 -4.954 -0.119 -0.145 1.00 66.55 C
HETATM 3 C MOL A 1 -4.943 1.171 -1.606 1.00 65.56 C
HETATM 4 C MOL A 1 -4.370 3.483 1.896 1.00 90.91 C
HETATM 5 C MOL A 1 -3.453 -1.004 5.509 1.00 73.85 C
HETATM 6 C MOL A 1 -3.173 4.220 -3.725 1.00 76.83 C
HETATM 7 C MOL A 1 -2.859 -2.921 3.533 1.00 73.72 C
HETATM 8 C MOL A 1 -2.662 1.621 -4.339 1.00 77.09 C
HETATM 9 C MOL A 1 -2.184 0.512 5.727 1.00 72.18 C
HETATM 10 C MOL A 1 -1.858 0.190 2.412 1.00 67.20 C
HETATM 11 C MOL A 1 -1.812 2.159 -1.115 1.00 69.06 C
HETATM 12 C MOL A 1 -0.430 -2.889 -2.034 1.00 48.57 C
HETATM 13 C MOL A 1 0.445 1.946 1.923 1.00 72.20 C
HETATM 14 C MOL A 1 1.001 2.017 -2.111 1.00 70.33 C
HETATM 15 C MOL A 1 1.204 1.859 5.826 1.00 77.88 C
HETATM 16 C MOL A 1 1.448 3.427 4.917 1.00 79.99 C
HETATM 17 C MOL A 1 1.629 -3.290 -0.464 1.00 49.22 C
HETATM 18 C MOL A 1 1.764 0.330 4.609 1.00 79.73 C
HETATM 19 C MOL A 1 1.684 5.181 2.383 1.00 83.67 C
HETATM 20 C MOL A 1 1.839 5.379 0.138 1.00 92.78 C
HETATM 21 C MOL A 1 2.158 5.259 -2.063 1.00 83.23 C
HETATM 22 C MOL A 1 2.792 3.626 -4.648 1.00 78.13 C
HETATM 23 C MOL A 1 3.092 0.538 -4.402 1.00 77.11 C
HETATM 24 C MOL A 1 3.696 2.173 2.330 1.00 69.55 C
HETATM 25 C MOL A 1 4.115 1.755 0.378 1.00 70.43 C
HETATM 26 C MOL A 1 4.228 2.323 -1.586 1.00 68.66 C
HETATM 1 O MOL A 1 -6.724 -3.149 -1.899 1.00 21.96 O
HETATM 2 O MOL A 1 -5.662 0.214 2.180 1.00 57.66 O
HETATM 3 O MOL A 1 -5.622 1.654 5.921 1.00 59.35 O
HETATM 4 O MOL A 1 -5.633 1.916 -0.962 1.00 58.44 O
HETATM 5 O MOL A 1 -5.543 6.017 -1.844 1.00 60.39 O
HETATM 6 O MOL A 1 -5.140 -1.881 2.703 1.00 54.52 O
HETATM 7 O MOL A 1 -5.102 1.437 -3.040 1.00 56.01 O
HETATM 8 O MOL A 1 -1.862 1.141 2.725 1.00 63.47 O
HETATM 9 O MOL A 1 -1.851 2.914 -0.340 1.00 65.00 O
HETATM 10 O MOL A 1 -1.336 -5.830 0.171 1.00 20.41 O
HETATM 11 O MOL A 1 -1.349 -1.099 3.164 1.00 59.42 O
HETATM 12 O MOL A 1 -1.348 2.393 -2.314 1.00 62.72 O
HETATM 13 O MOL A 1 -1.257 -3.039 -4.963 1.00 25.03 O
HETATM 14 O MOL A 1 0.050 1.303 3.049 1.00 65.30 O
HETATM 15 O MOL A 1 0.402 -2.696 -0.165 1.00 39.73 O
HETATM 16 O MOL A 1 0.976 1.380 -3.483 1.00 62.21 O
HETATM 17 O MOL A 1 2.317 -4.426 2.936 1.00 23.54 O
HETATM 18 O MOL A 1 3.118 -4.192 -3.021 1.00 24.16 O
HETATM 19 O MOL A 1 3.832 2.165 3.812 1.00 58.69 O
HETATM 20 O MOL A 1 4.227 -2.764 0.636 1.00 46.56 O
HETATM 21 O MOL A 1 4.751 2.471 -3.035 1.00 57.62 O
HETATM 22 O MOL A 1 7.276 -0.857 0.830 1.00 22.79 O
END
#############################
FYI, I optimized the structure in B3LYP/LANL2DZ/6-31G* and by the optimized structure again I obtained the .fchk file with all electron basis set DZP-DKH.
Could you make a clarification.
Thanks
Reza
#9Multiwfn and wavefunction analysis»2D ELF + AIM»2021-11-05 07:54:00
- rkia
- Replies: 1
-
Dear Prof. Lu,
Is there any possibility in multiwfn to combine the results of AIM and two dimensional ELF data together in one picture. I know how to combine 3D ELF data + NCI.
Kind regards,
Reza
#10Multiwfn and wavefunction analysis»Special RDG isosurface map»2021-09-13 16:02:19
- rkia
- Replies: 1
-
Dear Prof. Tian Lu,
I have seen a special RDG isosurface map in some published papers and they mentioned they have drawn by multiwfn program. I have seen different option but I couldn't find that in multiwfn. Could you please help me by commenting in this case.
I attached the image. The left side plot is very easy for me but I would like to have the right hand side plot complementary to the left one (LAPLACIAN).
With kind regards
Reza
#11Multiwfn and wavefunction analysis»AIM+NCI plot»2021-07-13 15:07:19
- rkia
- Replies: 1
-
Dear Prof. Tian Lu,
I know how to make AIM+NCI plot as you explained in your video. I would like to know if we can remove unwanted NCI interactions and just keep those isosurface between the atoms with critical point in the AIM+NCI plot.
Sometimes the combination of NCI plot along with the AIM is crowded.
Regards,
Reza
#12Multiwfn and wavefunction analysis»Deleting some intermolecular bond paths in VMD»2021-06-09 07:34:07
- rkia
- Replies: 1
-
Dear Sobereva,
When the interaction between two molecules shows different intermolecular bond paths, the graph will be complicated in VMD. I know how to remove unwanted critical points but I would like to know how I can remove unwanted bond paths and just show the important one.
Regards,
RK
#13Multiwfn and wavefunction analysis»bond path in VMD»2021-06-08 19:40:55
- rkia
- Replies: 1
-
Hi there,
How to remove unwanted paths in AIM plot visualized By VMD and keep the important one.
Regards
RK
#14Multiwfn and wavefunction analysis»RDGScatter coloured map»2021-03-02 03:09:11
- rkia
- Replies: 1
-
Dear Prof. Lu,
Based on your manual version 3.8. I generated output file of the NCI interactions and copied the output.txt and REDscatter.gnu script in the bin folder of gnuplot and tried to generate RDGscatter.ps file but the command "gnuplot RDGscatter.gnu" doesn't generate "RDGscatter.ps" file. I have installed irfanviwe and gnuplot properly. This is the Error message in DOS file:
Please help me to resolve this error. When I try to run this via command line of gnuplot, again it shows invalid command.

With kind regards,
Reza
Pages:1