Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » GNUplot command prompt » 2023-09-30 13:25:29
Sir, After doing IRI analysis I obtain output .txt then copying IRIscatter.gnu to Gnuplot. then running gnuplot IRIscatter.gnu in GNUPLOT folder. I found gnuplot iriscatter.gnu
^
invalid command
What to do to view IRIscatter .ps.
Yours sincerely,
Partha
#2 Re: Multiwfn and wavefunction analysis » Prepare useful batch script file » 2023-09-18 15:21:52
Sir, As per your instruction in Multiwfn mannual(4.2.5) you specify the VMD.bat file for easy plot high quality AIM topology in VMD visualization. But the VMD.bat is not found in script. Will you help.
Yours sincerely,
Partha
#3 Re: Multiwfn and wavefunction analysis » GENERATING FCH FILE » 2023-04-19 17:11:22
I have generated the fch file with a specific name. But during opening of the file I am unable to open. Please tell me the step wise , so that I can start the Multiwfn.
#4 Re: Multiwfn and wavefunction analysis » Electro density » 2020-08-30 06:36:41
Sir, I am able to run the problem changing the "nthread" 4 to 1.
Partha
#5 Re: Multiwfn and wavefunction analysis » Electro density » 2020-08-27 18:16:30
Sir, I have downloaded Multiwfn_3.7_bin_Win32. and tried to open Gaussian fch file as"C:/G09w/scratch/11chlorine.fch. But the program shows error
as C:/g09w/scratch/11chlorine.fch
Please wait...
Loading various information of the wavefunction
The highest angular moment basis functions is F
Loading basis set definition...
Loading orbitals...
Converting basis function information to GTF information...
Back converting basis function information from Cartesian to spherical type...
Generating density matrix based on SCF orbitals...
OMP: Error #136: Cannot create thread.
OMP: System error #8: Not enough storage is available to process this command.
OMP: Error #178: Function GetExitCodeThread() failed:
OMP: System error #6: The handle is invalid.
I am using Windows 32 system.
How can I solve the problem?
Partha
#6 Re: Multiwfn and wavefunction analysis » Electro density » 2020-08-23 15:20:27
Sir, For Cu complex binding to N and O have electron density of 0.0623 and 0.0580 e rspectively. What is the reason for more electron density (total) of nitrogen than oxygen in Multiwfn analysis?
Partha
#7 Re: Multiwfn and wavefunction analysis » Electro density » 2020-08-13 08:49:07
Sir, How can I obtained the figure like the following in topology analysis?
With regards,
Partha
letter2.doc
#8 Re: Multiwfn and wavefunction analysis » Electro density » 2020-08-07 08:15:41
Sir, For a complex like (drawn in figure), I found two types of data for charge decomposition.
CDA and ECDA data are given. I found from CDA data that Cl donates electron to the aromatic ring by [d - b = -0.195824 ] electron. On the other hand, ECDA result shows
CT( 1-> 2) - CT( 2-> 1) for all electrons: 0.0732. i.e, ring donates electron to electronegative Cl atom. Fragment one is the aromatic ring (-one Cl) and fragment 2 is Cl atom.
Which data is correct?
Waiting for reply, Partha
letter.doc
============= Charge decomposition analysis (CDA) result =============
d = The number of electrons donated from fragment 1 to fragment 2
b = The number of electrons back donated from fragment 2 to fragment 1
r = The number of electrons involved in repulsive polarization
**** Result for alpha electrons ****
Result for all electrons:
d= -0.042429 b= 0.153395 d - b = -0.195824 r= 0.018996
========== Extended Charge decomposition analysis (ECDA) ==========
**** Result for alpha electrons ****
Contribution to all occupied complex orbital:
Occupied, virtual orbitals of fragment 1: 10419.5844% 26.7444%
Occupied, virtual orbitals of fragment 2: 756.8508% 96.8204%
Contribution to all virtual complex orbital:
Occupied, virtual orbitals of fragment 1: 80.4156% 33173.2556%
Occupied, virtual orbitals of fragment 2: 43.1492% 903.1796%
PL( 1) + CT( 1-> 2) = 0.8042 PL( 1) + CT( 2-> 1) = 0.2674
PL( 2) + CT( 1-> 2) = 0.9682 PL( 2) + CT( 2-> 1) = 0.4315
The net electrons obtained by frag. 2 = CT( 1-> 2) - CT( 2-> 1) = 0.5367
**** Result for beta electrons ****
Contribution to all occupied complex orbital:
Occupied, virtual orbitals of fragment 1: 10465.3433% 81.0101%
Occupied, virtual orbitals of fragment 2: 853.1465% 0.5001%
Contribution to all virtual complex orbital:
Occupied, virtual orbitals of fragment 1: 34.6567% 33118.9899%
Occupied, virtual orbitals of fragment 2: 46.8535% 899.4999%
PL( 1) + CT( 1-> 2) = 0.3466 PL( 1) + CT( 2-> 1) = 0.8101
PL( 2) + CT( 1-> 2) = 0.0050 PL( 2) + CT( 2-> 1) = 0.4685
The net electrons obtained by frag. 2 = CT( 1-> 2) - CT( 2-> 1) = -0.4635
CT( 1-> 2) - CT( 2-> 1) for all electrons: 0.0732
#9 Re: Multiwfn and wavefunction analysis » Electro density » 2020-08-03 13:54:22
Sir, My desktop is of Windows 32 bit. But the new version is compatible with windows 64bit. My computing system does not support Multiwfn_3.7_dev_bin_win64.rar. How can I solve this?
Sincerely,
Partha
#10 Re: Multiwfn and wavefunction analysis » Electro density » 2020-08-02 16:01:47
sir, For a QTAIM ANALYSIS the following line appears,
"Iterative Hirshfeld (Hirshfeld-I) ===============
-2 Switch algorithm, current: Fast & large memory requirement
-1 Switch if output intermediate results, current: No
0 Return
1 Start calculation!
2 Set the maximum number of iterations, current: 50
3 Set convergence criterion of atomic charges, current: 0.000200
1
Could not find Gaussian path defined in "gaupath" variable in settings.ini
Input the path of Gaussian executable file, e.g. "D:\study\g09w\g09.exe"
1
Could not find Gaussian executable file, input again
How can I get this path (D:\study\g09w\g09.exe)?
yours Sincerely
Partha
#11 Re: Multiwfn and wavefunction analysis » Electro density » 2020-07-27 15:15:32
To
Prof. Tian Lu
Sir,
I am doing some charge decomposition analysis to study the ionophoric properties of the following complexes [ Fig 1] by Multiwfn_3.6_bin_win32 program.
I consider 3 units for all the complexes.
[1] [C9H4FNOCl]-1 (charge -1, multiplicity 1),
Unit
[2] [C9H4FNOCl]-1 ((charge -1, multiplicity 1)
and
unit [3] Cu2+(multiplicity 2).
I am using B3LYP protocol with cc-PVDZ basis set for F, Cl, Br and LANL2DZ for iodine. All the data shows almost same values of (d – b) [ 0.345458 ]. I suppose, I am doing something wrong as (d – b) values should be different for the 4 halogens situated at the 7 position of the ring ( nearest O donor). Please help me to do the charge decomposition analysis.
#12 Re: Multiwfn and wavefunction analysis » Electro density » 2020-07-20 05:44:46
Sir, I have to calculate charge decomposition of C6H5Cl using Multiwfn. For the total molecule charge is 0 with multiplicity 1. Now what will be the charge and multiplicity for "C6H5" fragment and "Cl" fragment?
#13 Re: Multiwfn and wavefunction analysis » Electro density » 2020-06-28 13:42:38
Sir, For a metal complex involving Cu[N,O donor], [Chlorine and fluorine atoms are inthe benzene ring]. I found that cu has0.96091 charges( Natural Population analysis) while Mulliken atomic chargeis 0.457333. On the same process N has -0.53717 and -0.29941 respectively. Whatis the reason behind this? What will be the better choice to represent?
Partha Sengupta
#14 Re: Multiwfn and wavefunction analysis » Electro density » 2020-06-04 05:51:37
Sir, I am able to draw the NBOs. But unable to determine the overlap region.Please help.
Partha
#15 Re: Multiwfn and wavefunction analysis » Electro density » 2020-06-04 05:18:25
As per your reply inCCL against my question, I am not able to draw the intercating NBO. I am able to see the MOs.with this following result.
Multiwfn -- A Multifunctional Wavefunction Analyzer
Version 3.6, release date: 2019-May-21
Project leader: Tian Lu (Beijing Kein Research Center for Natural Sciences)
Below paper *MUST BE CITED* if Multiwfn is utilized in your work:
Tian Lu, Feiwu Chen, J. Comput. Chem., 33, 580-592 (2012)
Multiwfn official website: //www.umsyar.com/multiwfn
Multiwfn English forum: //www.umsyar.com/wfnbbs
Multiwfn Chinese forum: http://bbs.keinsci.com/wfn
( The number of threads: 2 Current date: 2020-06-04 Time: 10:45:04 )
Please wait...
Loading various information of the wavefunction
The highest angular moment basis functions is F
Loading basis set definition...
Loading orbitals...
Converting basis function information to GTF information...
Generating overlap matrix...
Back converting basis function information from Cartesian to spherical type...
Generating density matrix based on SCF orbitals...
Total/Alpha/Beta electrons: 227.0000 114.0000 113.0000
Net charge: 0.00000 Expected multiplicity: 2
Atoms: 35, Basis functions: 455, GTFs: 1284
Total energy: -3711.581015869350 Hartree, Virial ratio: 2.00289719
This is an unrestricted single-determinant wavefunction
Orbitals from 1 to 455 are alpha, from 1 to 114 are occupied
Orbitals from 456 to 910 are beta, from 456 to 568 are occupied
Title line of this file: 11111Chloroflurociloquinolnbo
Loaded C:\Users\Partha\Desktop\Multiwfn_3.6_bin_Win32\11111Chloroflurociloquino
lnbo.fch successfully!
Formula: H8 C18 N2 O2 F2 Cl2 Cu1
Molecule weight: 456.71712
************ Main function menu ************
0 Show molecular structure and view orbitals
1 Output all properties at a point
2 Topology analysis
3 Output and plot specific property in a line
4 Output and plot specific property in a plane
5 Output and plot specific property within a spatial region (calc. grid data)
6 Check & modify wavefunction
7 Population analysis and atomic charges
8 Orbital composition analysis
9 Bond order analysis
10 Plot total DOS, partial DOS, OPDOS, local DOS and photoelectron spectrum
11 Plot IR/Raman/UV-Vis/ECD/VCD/ROA spectrum
12 Quantitative analysis of molecular surface
13 Process grid data (No grid data is presented currently)
14 Adaptive natural density partitioning (AdNDP) analysis
15 Fuzzy atomic space analysis
16 Charge decomposition analysis (CDA) and extended CDA (ECDA)
17 Basin analysis
18 Electron excitation analysis
19 Orbital localization analysis
20 Visual study of weak interaction
21 Energy decomposition analysis
100 Other functions (Part1) 200 Other functions (Part2)
0
Nucleus list:
1(C ) --> Charge: 6.000000 x,y,z(Bohr): -7.635001 -1.246586 0.000000
2(C ) --> Charge: 6.000000 x,y,z(Bohr): -5.114860 -0.443513 0.000000
3(C ) --> Charge: 6.000000 x,y,z(Bohr): -4.748994 2.245374 0.000000
4(C ) --> Charge: 6.000000 x,y,z(Bohr): -6.761073 4.026757 0.000000
5(C ) --> Charge: 6.000000 x,y,z(Bohr): -9.240213 3.037809 0.000000
6(C ) --> Charge: 6.000000 x,y,z(Bohr): -9.676138 0.478711 0.000000
7(C ) --> Charge: 6.000000 x,y,z(Bohr): -6.112241 6.621076 0.000000
8(H ) --> Charge: 1.000000 x,y,z(Bohr): -11.601392 -0.221535 0.000000
9(C ) --> Charge: 6.000000 x,y,z(Bohr): -3.599002 7.326711 0.000000
10(C ) --> Charge: 6.000000 x,y,z(Bohr): -1.706951 5.446656 0.000000
11(H ) --> Charge: 1.000000 x,y,z(Bohr): -7.605906 8.027042 0.000000
12(H ) --> Charge: 1.000000 x,y,z(Bohr): -3.053884 9.302607 0.000000
13(H ) --> Charge: 1.000000 x,y,z(Bohr): 0.295769 5.894209 0.000000
14(N ) --> Charge: 7.000000 x,y,z(Bohr): -2.287453 3.010668 0.000000
15(O ) --> Charge: 8.000000 x,y,z(Bohr): -3.126364 -1.918878 0.000000
16(F ) --> Charge: 9.000000 x,y,z(Bohr): -11.200311 4.681640 0.000000
17(Cl) --> Charge: 17.000000 x,y,z(Bohr): -8.291817 -4.483806 0.000000
18(C ) --> Charge: 6.000000 x,y,z(Bohr): 7.635320 1.246466 -0.000000
19(C ) --> Charge: 6.000000 x,y,z(Bohr): 5.115075 0.443926 -0.000000
20(C ) --> Charge: 6.000000 x,y,z(Bohr): 4.748602 -2.245058 -0.000000
21(C ) --> Charge: 6.000000 x,y,z(Bohr): 6.760512 -4.026761 -0.000000
22(C ) --> Charge: 6.000000 x,y,z(Bohr): 9.239950 -3.038345 -0.000000
23(C ) --> Charge: 6.000000 x,y,z(Bohr): 9.676233 -0.479387 -0.000000
24(C ) --> Charge: 6.000000 x,y,z(Bohr): 6.111251 -6.620868 -0.000000
25(H ) --> Charge: 1.000000 x,y,z(Bohr): 11.601607 0.220530 -0.000000
26(C ) --> Charge: 6.000000 x,y,z(Bohr): 3.597826 -7.326123 -0.000000
27(C ) --> Charge: 6.000000 x,y,z(Bohr): 1.706085 -5.445898 -0.000000
28(H ) --> Charge: 1.000000 x,y,z(Bohr): 7.604640 -8.027123 -0.000000
29(H ) --> Charge: 1.000000 x,y,z(Bohr): 3.052461 -9.301962 -0.000000
30(H ) --> Charge: 1.000000 x,y,z(Bohr): -0.296664 -5.893128 0.000000
31(N ) --> Charge: 7.000000 x,y,z(Bohr): 2.287068 -3.009888 -0.000000
32(O ) --> Charge: 8.000000 x,y,z(Bohr): 3.126719 1.919334 -0.000000
33(F ) --> Charge: 9.000000 x,y,z(Bohr): 11.199637 -4.682724 -0.000000
34(Cl) --> Charge: 17.000000 x,y,z(Bohr): 8.293641 4.483169 -0.000000
35(Cu) --> Charge: 29.000000 x,y,z(Bohr): -0.000000 0.000177 0.000000
Range of alpha orbitals: 1 - 455 Range of Beta orbitals: 456 - 910
Note: Orbital 114 is alpha-HOMO, energy: -0.129366 a.u. -3.520217 eV
Orbital 568 is beta-HOMO, energy: -0.128982 a.u. -3.509787 eV
Orbital 115 is alpha-LUMO, energy: -0.141716 a.u. -3.856293 eV
Orbital 569 is beta-LUMO, energy: -0.208126 a.u. -5.663383 eV
HOMO-LUMO gap of alpha orbitals: -0.012351 a.u. -0.336076 eV
HOMO-LUMO gap of beta orbitals: -0.079143 a.u. -2.153596 eV
120 E(au/eV): 0.01118 0.3043 Occ: 0.000000 Typ: A
Note: A set of grid data presents in memory
************ Main function menu ************
0 Show molecular structure and view orbitals
1 Output all properties at a point
2 Topology analysis
3 Output and plot specific property in a line
4 Output and plot specific property in a plane
5 Output and plot specific property within a spatial region (calc. grid data)
6 Check & modify wavefunction
7 Population analysis and atomic charges
8 Orbital composition analysis
9 Bond order analysis
10 Plot total DOS, partial DOS, OPDOS, local DOS and photoelectron spectrum
11 Plot IR/Raman/UV-Vis/ECD/VCD/ROA spectrum
12 Quantitative analysis of molecular surface
13 Process grid data
14 Adaptive natural density partitioning (AdNDP) analysis
15 Fuzzy atomic space analysis
16 Charge decomposition analysis (CDA) and extended CDA (ECDA)
17 Basin analysis
18 Electron excitation analysis
19 Orbital localization analysis
20 Visual study of weak interaction
21 Energy decomposition analysis
100 Other functions (Part1) 200 Other functions (Part2)
How can I see the interacting NBOs?
Partha
#16 Re: Multiwfn and wavefunction analysis » Electro density » 2020-05-26 13:56:43
Sir, Thank you for your kind attention to my question.
With regards,
Partha
#17 Multiwfn and wavefunction analysis » Electro density » 2020-05-24 08:27:30
- partha
- Replies: 24
Sir,
I have done a topology analysis of Copper complex with nitrogen and oxygen donor. The structure and BCPare given in the figure. I have to do the calculate the bond strength of Cu-O and Cu-N bonds. The specified critical points are 4, 5, 6 and 7.
I have to determine the 1. electron density (ρ)
2. Laplacian density 2((ρ)
3.electronic energy density(H9r)
4. electronic kinetic density (G(r)
5. potential energy density [V(r)] to specify the Cu-O and Cu-N bond.
How can I proceed to do the following calculation for two Cu-O bond and Cu-N bond. I have given the output file. Please help.
partha
The message I found
is given below
I have determine to
43 4.222221952 -4.777379338 0.000000000 (3,+1)
44 -1.309050536 -4.082445091 0.000000000 (3,-1)
45 -0.986040947 -3.815663713 0.000000000 (3,+1)
46 -7.916463547 -2.630315746 0.000000000 (3,-1)
47 7.988996985 -3.566256296 0.000000000 (3,-1)
48 5.766124289 -3.145784100 0.000000000 (3,-1)
49 9.880667714 -3.585449838 0.000000000 (3,-1)
50 3.909101992 -2.534195422 0.000000000 (3,-1)
51 -6.353852445 -0.870033413 0.000000000 (3,-1)
52 -4.447789083 -0.925519777 0.000000000 (3,-1)
53 -8.677057253 -0.373434694 0.000000000 (3,-1)
54 1.093250160 -1.448631766 0.000000000 (3,-1)
55 -10.923695979 0.023040313 0.000000000 (3,-1)
56 -1.549644007 -0.896915984 0.000000000 (3,-1)
57 9.478380715 -1.712727506 0.000000000 (3,-1)
58 7.181538052 -1.353067278 0.000000000 (3,+1)
59 4.932115914 -0.879535707 0.000000000 (3,-1)
60 2.720383244 -0.572031357 0.000000000 (3,+1)
61 -2.720303011 0.572667707 0.000000000 (3,+1)
62 -4.932178898 0.879924392 0.000000000 (3,-1)
63 -7.181666889 1.352961502 0.000000000 (3,+1)
64 -9.478444714 1.712131537 0.000000000 (3,-1)
65 1.549800214 0.897289869 0.000000000 (3,-1)
66 10.923863741 -0.023921339 0.000000000 (3,-1)
67 -1.093381928 1.449149648 0.000000000 (3,-1)
68 8.677220734 0.373036085 0.000000000 (3,-1)
69 4.448065171 0.925946767 0.000000000 (3,-1)
70 6.354131307 0.870202597 0.000000000 (3,-1)
71 -3.909427249 2.534700030 0.000000000 (3,-1)
72 -9.881070285 3.584722487 0.000000000 (3,-1)
73 -5.766603730 3.145944447 0.000000000 (3,-1)
74 -7.989396794 3.566004447 0.000000000 (3,-1)
75 7.917399245 2.629955934 0.000000000 (3,-1)
76 0.986732798 3.817205244 0.000000000 (3,+1)
77 1.308246429 4.082800089 0.000000000 (3,-1)
78 -4.222962305 4.777818397 0.000000000 (3,+1)
79 -1.927481580 4.635640546 0.000000000 (3,-1)
80 -6.442984445 5.325455984 0.000000000 (3,-1)
81 -0.389528074 5.744461508 0.000000000 (3,-1)
82 -2.676251291 6.402726706 0.000000000 (3,-1)
83 -4.841693921 6.976277560 0.000000000 (3,-1)
84 -7.081561687 7.533722350 0.000000000 (3,-1)
85 -3.246240603 8.600061906 0.000000000 (3,-1)
The number of critical points of each type:
(3,-3): 35, (3,-1): 42, (3,+1): 8, (3,+3): 0
Poincare-Hopf relationship verification: 35 - 42 + 8 - 0 = 1
Fine, Poincare-Hopf relationship is satisfied, all CPs may have been found
================ Topology analysis ===============
-11 Delete results and reselect real space function, current: 1
-10 Return
-9 Measure distances, angles and dihedral angles between CPs or atoms
-5 Modify or print detail or export paths, or plot property along a path
-4 Modify or export CPs (critical points)
-3 Set interbasin surface generating parameters
-2 Set path generating parameters
-1 Set CP searching parameters
0 Print and visualize all generated CPs, paths and interbasin surfaces
1 Search CPs from a given starting point
2 Search CPs from nuclear positions
3 Search CPs from midpoint of atom pairs
4 Search CPs from triangle center of three atoms
5 Search CPs from pyramid center of four atoms
6 Search CPs from a batch of points within a sphere
7 Show real space function values at specific CP or all CPs
8 Generating the paths connecting (3,-3) and (3,-1) CPs
9 Generating the paths connecting (3,+1) and (3,+3) CPs
10 Add or delete interbasin surfaces
20 Calculate Shannon aromaticity index
21 Calculate density curvature perpendicular to a specific plane at a point
7
Input the index of the CP that you are interested in, e.g. 3
Note 1: If input 0, then properties of all CPs will be outputted to CPprop.txt
in current folder (and if you feel the output speed is slow, you can input -1 to
avoid outputting ESP, which is the most expensive one)
Note 2: If input CP index with "d" suffix, e.g. 17d, then property of this CP c
an be decomposed into orbital contribution
7
Note: Unless otherwise specified, all units are in a.u.
CP Position: 1.70608714742820 -5.44589500480140 0.00000000000000
CP type: (3,-3)
Density of all electrons: 0.1180748351E+03
Density of Alpha electrons: 0.5903711047E+02
Density of Beta electrons: 0.5903772465E+02
Spin density of electrons: -0.6141774725E-03
Lagrangian kinetic energy G(r): 0.5123621072E+01
G(r) in X,Y,Z: 0.1989249274E+01 0.1758411109E+01 0.1375960689E+01
Hamiltonian kinetic energy K(r): 0.1091184750E+06
Potential energy density V(r): -0.1091235986E+06
Energy density E(r) or H(r): -0.1091184750E+06
Laplacian of electron density: -0.4364534056E+06
Electron localization function (ELF): 0.9999996057E+00
Localized orbital locator (LOL): 0.9993724617E+00
Local information entropy: 0.3311420907E+00
Reduced density gradient (RDG): 0.1000000000E+03
Reduced density gradient with promolecular approximation: 0.1000000000E+03
Sign(lambda2)*rho: -0.1180748351E+03
Sign(lambda2)*rho with promolecular approximation: -0.1213911689E+03
Corr. hole for alpha, ref.: 0.00000 0.00000 0.00000 : -0.3238987748E+00
Source function, ref.: 0.00000 0.00000 0.00000 : 0.6085960170E+04
Wavefunction value for orbital 1 : -0.1353653559E-04
Average local ionization energy (ALIE): 0.9916234739E+01
Delta_g: 0.4784170260E-01
User-defined real space function: 0.1000000000E+01
ESP from nuclear charges: 0.1456300882E+07
ESP from electrons: -0.4349775012E+02
Total ESP: 0.1456257385E+07 a.u. ( 0.3962678E+08 eV, 0.9138161E+09 kcal/mol)
Note: Below information are for electron density
Components of gradient in x/y/z are:
-0.1549624318E-10 -0.7426753657E-11 0.0000000000E+00
Norm of gradient is: 0.1718401066E-10
Components of Laplacian in x/y/z are:
-0.1454835838E+06 -0.1454844028E+06 -0.1454854189E+06
Total: -0.4364534056E+06
Hessian matrix:
-0.1454835838E+06 -0.9964115699E-01 0.0000000000E+00
-0.9964115699E-01 -0.1454844028E+06 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 -0.1454854189E+06
Eigenvalues of Hessian: -0.1454835719E+06 -0.1454844148E+06 -0.1454854189E+06
Eigenvectors(columns) of Hessian:
0.9928874646E+00 0.1190566359E+00 0.0000000000E+00
-0.1190566359E+00 0.9928874646E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.1000000000E+01
Determinant of Hessian: -0.3079285064E+16
Ellipticity of electron density: 0.000007
eta index: -1.000013
================ Topology analysis ===============
-11 Delete results and reselect real space function, current: 1
-10 Return
-9 Measure distances, angles and dihedral angles between CPs or atoms
-5 Modify or print detail or export paths, or plot property along a path
-4 Modify or export CPs (critical points)
-3 Set interbasin surface generating parameters
-2 Set path generating parameters
-1 Set CP searching parameters
0 Print and visualize all generated CPs, paths and interbasin surfaces
1 Search CPs from a given starting point
2 Search CPs from nuclear positions
3 Search CPs from midpoint of atom pairs
4 Search CPs from triangle center of three atoms
5 Search CPs from pyramid center of four atoms
6 Search CPs from a batch of points within a sphere
7 Show real space function values at specific CP or all CPs
8 Generating the paths connecting (3,-3) and (3,-1) CPs
9 Generating the paths connecting (3,+1) and (3,+3) CPs
10 Add or delete interbasin surfaces
20 Calculate Shannon aromaticity index
21 Calculate density curvature perpendicular to a specific plane at a point
0
No paths have been found
Summary of found CPs:
Index XYZ Coordinate (Bohr) Type
1 3.061874749 -9.267565181 0.000000000 (3,-3) Nuc: 29(H )
2 7.578278491 -8.002481393 0.000000000 (3,-3) Nuc: 28(H )
3 3.597826150 -7.326122186 0.000000000 (3,-3) Nuc: 26(C )
4 -0.260010257 -5.885435008 0.000000000 (3,-3) Nuc: 30(H )
5 6.111250960 -6.620866960 0.000000000 (3,-3) Nuc: 24(C )
6 -8.291816716 -4.483804737 0.000000000 (3,-3) Nuc: 17(Cl)
7 1.706087147 -5.445895005 0.000000000 (3,-3) Nuc: 27(C )
8 11.199631505 -4.682719099 0.000000000 (3,-3) Nuc: 33(F )
9 6.760511566 -4.026760814 0.000000000 (3,-3) Nuc: 21(C )
10 2.287072610 -3.009892628 0.000000000 (3,-3) Nuc: 31(N )
11 -3.126368653 -1.918871070 0.000000000 (3,-3) Nuc: 15(O )
12 -7.635002568 -1.246590925 0.000000000 (3,-3) Nuc: 1(C )
13 9.239956013 -3.038350438 0.000000000 (3,-3) Nuc: 22(C )
14 4.748598682 -2.245059796 0.000000000 (3,-3) Nuc: 20(C )
15 -11.567474636 -0.209274638 0.000000000 (3,-3) Nuc: 8(H )
16 -5.114857278 -0.443514888 0.000000000 (3,-3) Nuc: 2(C )
17 -9.676136871 0.478710926 0.000000000 (3,-3) Nuc: 6(C )
18 0.000000000 0.000177206 0.000000000 (3,-3) Nuc: 35(Cu)
19 9.676231211 -0.479386644 0.000000000 (3,-3) Nuc: 23(C )
20 5.115073009 0.443928222 0.000000000 (3,-3) Nuc: 19(C )
21 11.567687290 0.208276776 0.000000000 (3,-3) Nuc: 25(H )
22 -4.748990941 2.245375797 0.000000000 (3,-3) Nuc: 3(C )
23 -9.240219176 3.037814855 0.000000000 (3,-3) Nuc: 5(C )
24 7.635322171 1.246470863 0.000000000 (3,-3) Nuc: 18(C )
25 3.126723224 1.919326990 0.000000000 (3,-3) Nuc: 32(O )
26 -2.287458291 3.010672878 0.000000000 (3,-3) Nuc: 14(N )
27 -6.761073007 4.026756575 0.000000000 (3,-3) Nuc: 4(C )
28 -11.200305904 4.681635270 0.000000000 (3,-3) Nuc: 16(F )
29 -1.706952776 5.446652816 0.000000000 (3,-3) Nuc: 10(C )
30 8.293641006 4.483168477 0.000000000 (3,-3) Nuc: 34(Cl)
31 -6.112240431 6.621074869 0.000000000 (3,-3) Nuc: 7(C )
32 0.259119011 5.886511435 0.000000000 (3,-3) Nuc: 13(H )
33 -3.599002339 7.326709756 0.000000000 (3,-3) Nuc: 9(C )
34 -7.579539498 8.002405240 0.000000000 (3,-3) Nuc: 11(H )
35 -3.063293679 9.268208257 0.000000000 (3,-3) Nuc: 12(H )
36 3.244907497 -8.599436264 0.000000000 (3,-1)
37 7.080391077 -7.533702674 0.000000000 (3,-1)
38 4.840616241 -6.975873622 0.000000000 (3,-1)
39 2.675233026 -6.402054994 0.000000000 (3,-1)
40 0.388625850 -5.743488060 0.000000000 (3,-1)
41 6.442210169 -5.325345196 0.000000000 (3,-1)
42 1.926773078 -4.634859979 0.000000000 (3,-1)
43 4.222221952 -4.777379338 0.000000000 (3,+1)
44 -1.309050536 -4.082445091 0.000000000 (3,-1)
45 -0.986040947 -3.815663713 0.000000000 (3,+1)
46 -7.916463547 -2.630315746 0.000000000 (3,-1)
47 7.988996985 -3.566256296 0.000000000 (3,-1)
48 5.766124289 -3.145784100 0.000000000 (3,-1)
49 9.880667714 -3.585449838 0.000000000 (3,-1)
50 3.909101992 -2.534195422 0.000000000 (3,-1)
51 -6.353852445 -0.870033413 0.000000000 (3,-1)
52 -4.447789083 -0.925519777 0.000000000 (3,-1)
53 -8.677057253 -0.373434694 0.000000000 (3,-1)
54 1.093250160 -1.448631766 0.000000000 (3,-1)
55 -10.923695979 0.023040313 0.000000000 (3,-1)
56 -1.549644007 -0.896915984 0.000000000 (3,-1)
57 9.478380715 -1.712727506 0.000000000 (3,-1)
58 7.181538052 -1.353067278 0.000000000 (3,+1)
59 4.932115914 -0.879535707 0.000000000 (3,-1)
60 2.720383244 -0.572031357 0.000000000 (3,+1)
61 -2.720303011 0.572667707 0.000000000 (3,+1)
62 -4.932178898 0.879924392 0.000000000 (3,-1)
63 -7.181666889 1.352961502 0.000000000 (3,+1)
64 -9.478444714 1.712131537 0.000000000 (3,-1)
65 1.549800214 0.897289869 0.000000000 (3,-1)
66 10.923863741 -0.023921339 0.000000000 (3,-1)
67 -1.093381928 1.449149648 0.000000000 (3,-1)
68 8.677220734 0.373036085 0.000000000 (3,-1)
69 4.448065171 0.925946767 0.000000000 (3,-1)
70 6.354131307 0.870202597 0.000000000 (3,-1)
71 -3.909427249 2.534700030 0.000000000 (3,-1)
72 -9.881070285 3.584722487 0.000000000 (3,-1)
73 -5.766603730 3.145944447 0.000000000 (3,-1)
74 -7.989396794 3.566004447 0.000000000 (3,-1)
75 7.917399245 2.629955934 0.000000000 (3,-1)
76 0.986732798 3.817205244 0.000000000 (3,+1)
77 1.308246429 4.082800089 0.000000000 (3,-1)
78 -4.222962305 4.777818397 0.000000000 (3,+1)
79 -1.927481580 4.635640546 0.000000000 (3,-1)
80 -6.442984445 5.325455984 0.000000000 (3,-1)
81 -0.389528074 5.744461508 0.000000000 (3,-1)
82 -2.676251291 6.402726706 0.000000000 (3,-1)
83 -4.841693921 6.976277560 0.000000000 (3,-1)
84 -7.081561687 7.533722350 0.000000000 (3,-1)
85 -3.246240603 8.600061906 0.000000000 (3,-1)
The number of critical points of each type:
(3,-3): 35, (3,-1): 42, (3,+1): 8, (3,+3): 0
Poincare-Hopf relationship verification: 35 - 42 + 8 - 0 = 1
Fine, Poincare-Hopf relationship is satisfied, all CPs may have been found
================ Topology analysis ===============
-11 Delete results and reselect real space function, current: 1
-10 Return
-9 Measure distances, angles and dihedral angles between CPs or atoms
-5 Modify or print detail or export paths, or plot property along a path
-4 Modify or export CPs (critical points)
-3 Set interbasin surface generating parameters
-2 Set path generating parameters
-1 Set CP searching parameters
0 Print and visualize all generated CPs, paths and interbasin surfaces
1 Search CPs from a given starting point
2 Search CPs from nuclear positions
3 Search CPs from midpoint of atom pairs
4 Search CPs from triangle center of three atoms
5 Search CPs from pyramid center of four atoms
6 Search CPs from a batch of points within a sphere
7 Show real space function values at specific CP or all CPs
8 Generating the paths connecting (3,-3) and (3,-1) CPs
9 Generating the paths connecting (3,+1) and (3,+3) CPs
10 Add or delete interbasin surfaces
20 Calculate Shannon aromaticity index
21 Calculate density curvature perpendicular to a specific plane at a point
7
Input the index of the CP that you are interested in, e.g. 3
Note 1: If input 0, then properties of all CPs will be outputted to CPprop.txt
in current folder (and if you feel the output speed is slow, you can input -1 to
avoid outputting ESP, which is the most expensive one)
Note 2: If input CP index with "d" suffix, e.g. 17d, then property of this CP c
an be decomposed into orbital contribution
7d
Select the function to be studied
----------- Avaliable real space functions -----------
1 Electron density 2 Gradient norm of electron density
3 Laplacian of electron density 4 Value of orbital wavefunction
5 Electron spin density
6 Hamiltonian kinetic energy density K(r)
7 Lagrangian kinetic energy density G(r)
8 Electrostatic potential from nuclear charges
9 Electron Localization Function (ELF)
10 Localized orbital locator (LOL)
11 Local information entropy
12 Total electrostatic potential (ESP)
13 Reduced density gradient (RDG) 14 RDG with promolecular approximation
15 Sign(lambda2)*rho 16 Sign(lambda2)*rho with promolecular approximation
17 Correlation hole for alpha, ref. point: 0.00000 0.00000 0.00000
18 Average local ionization energy (ALIE)
19 Source function, mode: 1, ref. point: 0.00000 0.00000 0.00000
20 Electron delocalization range function EDR(r;d)
21 Orbital overlap distance function D(r)
22 Delta_g function
100 User-defined real space function, iuserfunc= 0
============ 8 Generating the paths connecting (3,-3) and (3,-1) CPs
9 Generating the paths connecting (3,+1) and (3,+3) CPs
10 Add or delete interbasin surfaces
20 Calculate Shannon aromaticity index
21 Calculate density curvature perpendicular to a specific plane at a point
0
No paths have been found
Summary of found CPs:
Index XYZ Coordinate (Bohr) Type
1 3.061874749 -9.267565181 0.000000000 (3,-3) Nuc: 29(H )
2 7.578278491 -8.002481393 0.000000000 (3,-3) Nuc: 28(H )
3 3.597826150 -7.326122186 0.000000000 (3,-3) Nuc: 26(C )
4 -0.260010257 -5.885435008 0.000000000 (3,-3) Nuc: 30(H )
5 6.111250960 -6.620866960 0.000000000 (3,-3) Nuc: 24(C )
6 -8.291816716 -4.483804737 0.000000000 (3,-3) Nuc: 17(Cl)
7 1.706087147 -5.445895005 0.000000000 (3,-3) Nuc: 27(C )
8 11.199631505 -4.682719099 0.000000000 (3,-3) Nuc: 33(F )
9 6.760511566 -4.026760814 0.000000000 (3,-3) Nuc: 21(C )
10 2.287072610 -3.009892628 0.000000000 (3,-3) Nuc: 31(N )
11 -3.126368653 -1.918871070 0.000000000 (3,-3) Nuc: 15(O )
12 -7.635002568 -1.246590925 0.000000000 (3,-3) Nuc: 1(C )
13 9.239956013 -3.038350438 0.000000000 (3,-3) Nuc: 22(C )
14 4.748598682 -2.245059796 0.000000000 (3,-3) Nuc: 20(C )
15 -11.567474636 -0.209274638 0.000000000 (3,-3) Nuc: 8(H )
16 -5.114857278 -0.443514888 0.000000000 (3,-3) Nuc: 2(C )
17 -9.676136871 0.478710926 0.000000000 (3,-3) Nuc: 6(C )
18 0.000000000 0.000177206 0.000000000 (3,-3) Nuc: 35(Cu)
19 9.676231211 -0.479386644 0.000000000 (3,-3) Nuc: 23(C )
20 5.115073009 0.443928222 0.000000000 (3,-3) Nuc: 19(C )
21 11.567687290 0.208276776 0.000000000 (3,-3) Nuc: 25(H )
22 -4.748990941 2.245375797 0.000000000 (3,-3) Nuc: 3(C )
23 -9.240219176 3.037814855 0.000000000 (3,-3) Nuc: 5(C )
24 7.635322171 1.246470863 0.000000000 (3,-3) Nuc: 18(C )
25 3.126723224 1.919326990 0.000000000 (3,-3) Nuc: 32(O )
26 -2.287458291 3.010672878 0.000000000 (3,-3) Nuc: 14(N )
27 -6.761073007 4.026756575 0.000000000 (3,-3) Nuc: 4(C )
28 -11.200305904 4.681635270 0.000000000 (3,-3) Nuc: 16(F )
29 -1.706952776 5.446652816 0.000000000 (3,-3) Nuc: 10(C )
30 8.293641006 4.483168477 0.000000000 (3,-3) Nuc: 34(Cl)
31 -6.112240431 6.621074869 0.000000000 (3,-3) Nuc: 7(C )
32 0.259119011 5.886511435 0.000000000 (3,-3) Nuc: 13(H )
33 -3.599002339 7.326709756 0.000000000 (3,-3) Nuc: 9(C )
34 -7.579539498 8.002405240 0.000000000 (3,-3) Nuc: 11(H )
35 -3.063293679 9.268208257 0.000000000 (3,-3) Nuc: 12(H )
36 3.244907497 -8.599436264 0.000000000 (3,-1)
37 7.080391077 -7.533702674 0.000000000 (3,-1)
38 4.840616241 -6.975873622 0.000000000 (3,-1)
39 2.675233026 -6.402054994 0.000000000 (3,-1)
40 0.388625850 -5.743488060 0.000000000 (3,-1)
41 6.442210169 -5.325345196 0.000000000 (3,-1)
42 1.926773078 -4.634859979 0.000000000 (3,-1)
43 4.222221952 -4.777379338 0.000000000 (3,+1)
44 -1.309050536 -4.082445091 0.000000000 (3,-1)
45 -0.986040947 -3.815663713 0.000000000 (3,+1)
46 -7.916463547 -2.630315746 0.000000000 (3,-1)
47 7.988996985 -3.566256296 0.000000000 (3,-1)
48 5.766124289 -3.145784100 0.000000000 (3,-1)
49 9.880667714 -3.585449838 0.000000000 (3,-1)
50 3.909101992 -2.534195422 0.000000000 (3,-1)
51 -6.353852445 -0.870033413 0.000000000 (3,-1)
52 -4.447789083 -0.925519777 0.000000000 (3,-1)
53 -8.677057253 -0.373434694 0.000000000 (3,-1)
54 1.093250160 -1.448631766 0.000000000 (3,-1)
55 -10.923695979 0.023040313 0.000000000 (3,-1)
56 -1.549644007 -0.896915984 0.000000000 (3,-1)
57 9.478380715 -1.712727506 0.000000000 (3,-1)
58 7.181538052 -1.353067278 0.000000000 (3,+1)
59 4.932115914 -0.879535707 0.000000000 (3,-1)
60 2.720383244 -0.572031357 0.000000000 (3,+1)
61 -2.720303011 0.572667707 0.000000000 (3,+1)
62 -4.932178898 0.879924392 0.000000000 (3,-1)
63 -7.181666889 1.352961502 0.000000000 (3,+1)
64 -9.478444714 1.712131537 0.000000000 (3,-1)
65 1.549800214 0.897289869 0.000000000 (3,-1)
66 10.923863741 -0.023921339 0.000000000 (3,-1)
67 -1.093381928 1.449149648 0.000000000 (3,-1)
68 8.677220734 0.373036085 0.000000000 (3,-1)
69 4.448065171 0.925946767 0.000000000 (3,-1)
70 6.354131307 0.870202597 0.000000000 (3,-1)
71 -3.909427249 2.534700030 0.000000000 (3,-1)
72 -9.881070285 3.584722487 0.000000000 (3,-1)
73 -5.766603730 3.145944447 0.000000000 (3,-1)
74 -7.989396794 3.566004447 0.000000000 (3,-1)
75 7.917399245 2.629955934 0.000000000 (3,-1)
76 0.986732798 3.817205244 0.000000000 (3,+1)
77 1.308246429 4.082800089 0.000000000 (3,-1)
78 -4.222962305 4.777818397 0.000000000 (3,+1)
79 -1.927481580 4.635640546 0.000000000 (3,-1)
80 -6.442984445 5.325455984 0.000000000 (3,-1)
81 -0.389528074 5.744461508 0.000000000 (3,-1)
82 -2.676251291 6.402726706 0.000000000 (3,-1)
83 -4.841693921 6.976277560 0.000000000 (3,-1)
84 -7.081561687 7.533722350 0.000000000 (3,-1)
85 -3.246240603 8.600061906 0.000000000 (3,-1)
The number of critical points of each type:
(3,-3): 35, (3,-1): 42, (3,+1): 8, (3,+3): 0
Poincare-Hopf relationship verification: 35 - 42 + 8 - 0 = 1
Fine, Poincare-Hopf relationship is satisfied, all CPs may have been found
================ Topology analysis ===============
-11 Delete results and reselect real space function, current: 1
-10 Return
-9 Measure distances, angles and dihedral angles between CPs or atoms
-5 Modify or print detail or export paths, or plot property along a path
-4 Modify or export CPs (critical points)
-3 Set interbasin surface generating parameters
-2 Set path generating parameters
-1 Set CP searching parameters
0 Print and visualize all generated CPs, paths and interbasin surfaces
1 Search CPs from a given starting point
2 Search CPs from nuclear positions
3 Search CPs from midpoint of atom pairs
4 Search CPs from triangle center of three atoms
5 Search CPs from pyramid center of four atoms
6 Search CPs from a batch of points within a sphere
7 Show real space function values at specific CP or all CPs
8 Generating the paths connecting (3,-3) and (3,-1) CPs
9 Generating the paths connecting (3,+1) and (3,+3) CPs
10 Add or delete interbasin surfaces
20 Calculate Shannon aromaticity index
21 Calculate density curvature perpendicular to a specific plane at a point
7
Input the index of the CP that you are interested in, e.g. 3
Note 1: If input 0, then properties of all CPs will be outputted to CPprop.txt
in current folder (and if you feel the output speed is slow, you can input -1 to
avoid outputting ESP, which is the most expensive one)
Note 2: If input CP index with "d" suffix, e.g. 17d, then property of this CP c
an be decomposed into orbital contribution
7
Note: Unless otherwise specified, all units are in a.u.
CP Position: 1.70608714742820 -5.44589500480140 0.00000000000000
CP type: (3,-3)
Density of all electrons: 0.1180748351E+03
Density of Alpha electrons: 0.5903711047E+02
Density of Beta electrons: 0.5903772465E+02
Spin density of electrons: -0.6141774725E-03
Lagrangian kinetic energy G(r): 0.5123621072E+01
G(r) in X,Y,Z: 0.1989249274E+01 0.1758411109E+01 0.1375960689E+01
Hamiltonian kinetic energy K(r): 0.1091184750E+06
Potential energy density V(r): -0.1091235986E+06
Energy density E(r) or H(r): -0.1091184750E+06
Laplacian of electron density: -0.4364534056E+06
Electron localization function (ELF): 0.9999996057E+00
Localized orbital locator (LOL): 0.9993724617E+00
Local information entropy: 0.3311420907E+00
Reduced density gradient (RDG): 0.1000000000E+03
Reduced density gradient with promolecular approximation: 0.1000000000E+03
Sign(lambda2)*rho: -0.1180748351E+03
Sign(lambda2)*rho with promolecular approximation: -0.1213911689E+03
Corr. hole for alpha, ref.: 0.00000 0.00000 0.00000 : -0.3238987748E+00
Source function, ref.: 0.00000 0.00000 0.00000 : 0.6085960170E+04
Wavefunction value for orbital 1 : -0.1353653559E-04
Average local ionization energy (ALIE): 0.9916234739E+01
Delta_g: 0.4784170260E-01
User-defined real space function: 0.1000000000E+01
ESP from nuclear charges: 0.1456300882E+07
ESP from electrons: -0.4349775012E+02
Total ESP: 0.1456257385E+07 a.u. ( 0.3962678E+08 eV, 0.9138161E+09 kcal/mol)
Note: Below information are for electron density
Components of gradient in x/y/z are:
-0.1549624318E-10 -0.7426753657E-11 0.0000000000E+00
Norm of gradient is: 0.1718401066E-10
Components of Laplacian in x/y/z are:
-0.1454835838E+06 -0.1454844028E+06 -0.1454854189E+06
Total: -0.4364534056E+06
Hessian matrix:
-0.1454835838E+06 -0.9964115699E-01 0.0000000000E+00
-0.9964115699E-01 -0.1454844028E+06 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 -0.1454854189E+06
Eigenvalues of Hessian: -0.1454835719E+06 -0.1454844148E+06 -0.1454854189E+06
Eigenvectors(columns) of Hessian:
0.9928874646E+00 0.1190566359E+00 0.0000000000E+00
-0.1190566359E+00 0.9928874646E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.1000000000E+01
Determinant of Hessian: -0.3079285064E+16
Ellipticity of electron density: 0.000007
eta index: -1.000013
================ Topology analysis ===============
-11 Delete results and reselect real space function, current: 1
-10 Return
-9 Measure distances, angles and dihedral angles between CPs or atoms
-5 Modify or print detail or export paths, or plot property along a path
-4 Modify or export CPs (critical points)
-3 Set interbasin surface generating parameters
-2 Set path generating parameters
-1 Set CP searching parameters
0 Print and visualize all generated CPs, paths and interbasin surfaces
1 Search CPs from a given starting point
2 Search CPs from nuclear positions
3 Search CPs from midpoint of atom pairs
4 Search CPs from triangle center of three atoms
5 Search CPs from pyramid center of four atoms
6 Search CPs from a batch of points within a sphere
7 Show real space function values at specific CP or all CPs
8 Generating the paths connecting (3,-3) and (3,-1) CPs
9 Generating the paths connecting (3,+1) and (3,+3) CPs
10 Add or delete interbasin surfaces
20 Calculate Shannon aromaticity index
21 Calculate density curvature perpendicular to a specific plane at a point
0
No paths have been found
Summary of found CPs:
Index XYZ Coordinate (Bohr) Type
1 3.061874749 -9.267565181 0.000000000 (3,-3) Nuc: 29(H )
2 7.578278491 -8.002481393 0.000000000 (3,-3) Nuc: 28(H )
3 3.597826150 -7.326122186 0.000000000 (3,-3) Nuc: 26(C )
4 -0.260010257 -5.885435008 0.000000000 (3,-3) Nuc: 30(H )
5 6.111250960 -6.620866960 0.000000000 (3,-3) Nuc: 24(C )
6 -8.291816716 -4.483804737 0.000000000 (3,-3) Nuc: 17(Cl)
7 1.706087147 -5.445895005 0.000000000 (3,-3) Nuc: 27(C )
8 11.199631505 -4.682719099 0.000000000 (3,-3) Nuc: 33(F )
9 6.760511566 -4.026760814 0.000000000 (3,-3) Nuc: 21(C )
10 2.287072610 -3.009892628 0.000000000 (3,-3) Nuc: 31(N )
11 -3.126368653 -1.918871070 0.000000000 (3,-3) Nuc: 15(O )
12 -7.635002568 -1.246590925 0.000000000 (3,-3) Nuc: 1(C )
13 9.239956013 -3.038350438 0.000000000 (3,-3) Nuc: 22(C )
14 4.748598682 -2.245059796 0.000000000 (3,-3) Nuc: 20(C )
15 -11.567474636 -0.209274638 0.000000000 (3,-3) Nuc: 8(H )
16 -5.114857278 -0.443514888 0.000000000 (3,-3) Nuc: 2(C )
17 -9.676136871 0.478710926 0.000000000 (3,-3) Nuc: 6(C )
18 0.000000000 0.000177206 0.000000000 (3,-3) Nuc: 35(Cu)
19 9.676231211 -0.479386644 0.000000000 (3,-3) Nuc: 23(C )
20 5.115073009 0.443928222 0.000000000 (3,-3) Nuc: 19(C )
21 11.567687290 0.208276776 0.000000000 (3,-3) Nuc: 25(H )
22 -4.748990941 2.245375797 0.000000000 (3,-3) Nuc: 3(C )
23 -9.240219176 3.037814855 0.000000000 (3,-3) Nuc: 5(C )
24 7.635322171 1.246470863 0.000000000 (3,-3) Nuc: 18(C )
25 3.126723224 1.919326990 0.000000000 (3,-3) Nuc: 32(O )
26 -2.287458291 3.010672878 0.000000000 (3,-3) Nuc: 14(N )
27 -6.761073007 4.026756575 0.000000000 (3,-3) Nuc: 4(C )
28 -11.200305904 4.681635270 0.000000000 (3,-3) Nuc: 16(F )
29 -1.706952776 5.446652816 0.000000000 (3,-3) Nuc: 10(C )
30 8.293641006 4.483168477 0.000000000 (3,-3) Nuc: 34(Cl)
31 -6.112240431 6.621074869 0.000000000 (3,-3) Nuc: 7(C )
32 0.259119011 5.886511435 0.000000000 (3,-3) Nuc: 13(H )
33 -3.599002339 7.326709756 0.000000000 (3,-3) Nuc: 9(C )
34 -7.579539498 8.002405240 0.000000000 (3,-3) Nuc: 11(H )
35 -3.063293679 9.268208257 0.000000000 (3,-3) Nuc: 12(H )
36 3.244907497 -8.599436264 0.000000000 (3,-1)
37 7.080391077 -7.533702674 0.000000000 (3,-1)
38 4.840616241 -6.975873622 0.000000000 (3,-1)
39 2.675233026 -6.402054994 0.000000000 (3,-1)
40 0.388625850 -5.743488060 0.000000000 (3,-1)
41 6.442210169 -5.325345196 0.000000000 (3,-1)
42 1.926773078 -4.634859979 0.000000000 (3,-1)
43 4.222221952 -4.777379338 0.000000000 (3,+1)
44 -1.309050536 -4.082445091 0.000000000 (3,-1)
45 -0.986040947 -3.815663713 0.000000000 (3,+1)
46 -7.916463547 -2.630315746 0.000000000 (3,-1)
47 7.988996985 -3.566256296 0.000000000 (3,-1)
48 5.766124289 -3.145784100 0.000000000 (3,-1)
49 9.880667714 -3.585449838 0.000000000 (3,-1)
50 3.909101992 -2.534195422 0.000000000 (3,-1)
51 -6.353852445 -0.870033413 0.000000000 (3,-1)
52 -4.447789083 -0.925519777 0.000000000 (3,-1)
53 -8.677057253 -0.373434694 0.000000000 (3,-1)
54 1.093250160 -1.448631766 0.000000000 (3,-1)
55 -10.923695979 0.023040313 0.000000000 (3,-1)
56 -1.549644007 -0.896915984 0.000000000 (3,-1)
57 9.478380715 -1.712727506 0.000000000 (3,-1)
58 7.181538052 -1.353067278 0.000000000 (3,+1)
59 4.932115914 -0.879535707 0.000000000 (3,-1)
60 2.720383244 -0.572031357 0.000000000 (3,+1)
61 -2.720303011 0.572667707 0.000000000 (3,+1)
62 -4.932178898 0.879924392 0.000000000 (3,-1)
63 -7.181666889 1.352961502 0.000000000 (3,+1)
64 -9.478444714 1.712131537 0.000000000 (3,-1)
65 1.549800214 0.897289869 0.000000000 (3,-1)
66 10.923863741 -0.023921339 0.000000000 (3,-1)
67 -1.093381928 1.449149648 0.000000000 (3,-1)
68 8.677220734 0.373036085 0.000000000 (3,-1)
69 4.448065171 0.925946767 0.000000000 (3,-1)
70 6.354131307 0.870202597 0.000000000 (3,-1)
71 -3.909427249 2.534700030 0.000000000 (3,-1)
72 -9.881070285 3.584722487 0.000000000 (3,-1)
73 -5.766603730 3.145944447 0.000000000 (3,-1)
74 -7.989396794 3.566004447 0.000000000 (3,-1)
75 7.917399245 2.629955934 0.000000000 (3,-1)
76 0.986732798 3.817205244 0.000000000 (3,+1)
77 1.308246429 4.082800089 0.000000000 (3,-1)
78 -4.222962305 4.777818397 0.000000000 (3,+1)
79 -1.927481580 4.635640546 0.000000000 (3,-1)
80 -6.442984445 5.325455984 0.000000000 (3,-1)
81 -0.389528074 5.744461508 0.000000000 (3,-1)
82 -2.676251291 6.402726706 0.000000000 (3,-1)
83 -4.841693921 6.976277560 0.000000000 (3,-1)
84 -7.081561687 7.533722350 0.000000000 (3,-1)
85 -3.246240603 8.600061906 0.000000000 (3,-1)
The number of critical points of each type:
(3,-3): 35, (3,-1): 42, (3,+1): 8, (3,+3): 0
Poincare-Hopf relationship verification: 35 - 42 + 8 - 0 = 1
Fine, Poincare-Hopf relationship is satisfied, all CPs may have been found
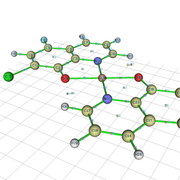
#18 Re: Multiwfn and wavefunction analysis » failure of Multiwfn work » 2020-05-08 16:53:54
Sir, I have done the calculation.Gaussian 09W. The input in Gaussian file is
"%rwf=9Chloroflurociloquinol.rwf
%chk=9Chloroflurociloquinol.chk
--------------------------------
# b3lyp/6-31g(d) pop=hirshfeldee
Molecule
.....
But during the calculation i find
"Multiwfn -- A Multifunctional Wavefunction Analyzer
Version 3.6, release date: 2019-May-21
Project leader: Tian Lu (Beijing Kein Research Center for Natural Sciences)
Below paper *MUST BE CITED* if Multiwfn is utilized in your work:
Tian Lu, Feiwu Chen, J. Comput. Chem., 33, 580-592 (2012)
Multiwfn official website: //www.umsyar.com/multiwfn
Multiwfn English forum: //www.umsyar.com/wfnbbs
Multiwfn Chinese forum: http://bbs.keinsci.com/wfn
( The number of threads: 2 Current date: 2020-05-08 Time: 21:56:26 )
Please wait...
Loading various information of the wavefunction
The highest angular moment basis functions is D
Loading basis set definition...
Loading orbitals...
Converting basis function information to GTF information...
Generating overlap matrix...
Generating density matrix based on SCF orbitals...
Total/Alpha/Beta electrons: 100.0000 50.0000 50.0000
Net charge: 0.00000 Expected multiplicity: 1
Atoms: 18, Basis functions: 209, GTFs: 408
Total energy: -1035.970153780311 Hartree, Virial ratio: 2.00629841
This is a restricted single-determinant wavefunction
Orbitals from 1 to 50 are occupied
Title line of this file: 9Chloroflurociloquinol
Loaded C:\Multiwfn_3.6_bin_Win32\9Chloroflurociloquinol.fch successfully!
Formula: H5 C9 N1 O1 F1 Cl1
Molecule weight: 197.59350
************ Main function menu ************
0 Show molecular structure and view orbitals
1 Output all properties at a point
2 Topology analysis
3 Output and plot specific property in a line
4 Output and plot specific property in a plane
5 Output and plot specific property within a spatial region (calc. grid data)
6 Check & modify wavefunction
7 Population analysis and atomic charges
8 Orbital composition analysis
9 Bond order analysis
10 Plot total DOS, partial DOS, OPDOS, local DOS and photoelectron spectrum
11 Plot IR/Raman/UV-Vis/ECD/VCD/ROA spectrum
12 Quantitative analysis of molecular surface
13 Process grid data (No grid data is presented currently)
14 Adaptive natural density partitioning (AdNDP) analysis
15 Fuzzy atomic space analysis
16 Charge decomposition analysis (CDA) and extended CDA (ECDA)
17 Basin analysis
18 Electron excitation analysis
19 Orbital localization analysis
20 Visual study of weak interaction
21 Energy decomposition analysis
100 Other functions (Part1) 200 Other functions (Part2)
7//
============== Population analysis ==============
-2 Calculate interaction energy between fragments based on atomic charges
-1 Define fragment
0 Return
1 Hirshfeld atom population
2 Voronoi deformation density (VDD) atom population
5 Mulliken atom & basis function population analysis
6 Lowdin atom & basis function population analysis
7 Modified Mulliken atom population defined by Ros & Schuit (SCPA)
8 Modified Mulliken atom population defined by Stout & Politzer
9 Modified Mulliken atom population defined by Bickelhaupt
10 Becke atomic charge with atomic dipole moment correction
11 Atomic dipole corrected Hirshfeld atomic charge (ADCH) (recommended)
12 CHELPG ESP fitting atomic charge
13 Merz-Kollmann (MK) ESP fitting atomic charge
14 AIM atomic charge
15 Hirshfeld-I atom population
16 CM5 atomic charge
17 Electronegativity Equalization Method (EEM) atomic charge
18 Restrained ElectroStatic Potential (RESP) atomic charge
1//
Citation: Theor. Chim. Acta. (Berl), 44, 129-138 (1977)
This task requests atomic densities, please select how to obtain them
1 Use build-in sphericalized atomic densities in free-states (more convenient)
2 Provide wavefunction file of involved elements by yourself or invoke Gaussian
to automatically calculate them
2
Running: rmdir /S /Q wfntmp
Running: mkdir wfntmp
Note: Some or all atom .wfn files needed are not present in "atomwfn" folder, t
hey must be calculated now. See Section 3.7.3 of the manual for detail.
Now please input the level for calculating atom wfn files, theoretical method i
s optional.
For example: 6-31G* or B3LYP/def2SVP You can also add other keywords at the
same time, e.g. M062X/6-311G(2d,p) scf=xqc int=ultrafine
6-31g*
Could not find Gaussian path defined in "gaupath" variable in settings.ini
Input the path of Gaussian executable file, e.g. "D:\study\g16w\g16.exe"
In this stage I am unable to follow the required instruction to define gaupath. I have asked Gaussian, they told me this may be the requirement of “Multiwfn”
=====================
After you select subfunctions -1 or -2 to study promolecular and deformation properties, Multiwfn checks whether .wfn files of all elements involved in present system have been presented in “atomwfn” subdirectory of current directory, if not, Multiwfn automatically invokes Gaussian to generate the missing element .wfn files and sphericalizes their densities. If the path of Gaussian executable file (“gaupath” parameter in settings.ini) is incorrect or has not been defined, Multiwfn will ask you to input the path of Gaussian executable file. (but how?)
Waiting for reply, With sincere thanks,
partha
#19 Multiwfn and wavefunction analysis » failure of Multiwfn work » 2020-05-07 15:46:28
- partha
- Replies: 3
I am newcomer. I am trying to use Multifwn for calculation. I tried to calculate density curvature to a specic poin. After all calculation the command prompt vanishes with no indication of data. What to do?
partha
Pages: 1