Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 Re: Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 13:23:49
Thanks. I got it. I watched the Youtube Video, and did as you explained. everything is OK.
#2 Re: Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 12:27:47
there are some script files (batch files if I am correct) in the YOUTUBE video. How can we get them?
#3 Re: Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 12:19:32
OK, I will test it. Thanks.
#4 Re: Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 10:07:25
It seems that only carbon atoms should be bounded. But how? GaussView automatically shows all the bonds.
#5 Re: Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 10:05:53
this is for the time that I uncheck the bonds:

#6 Re: Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 10:03:24
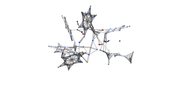
#7 Re: Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 09:58:18
Hi again
I used -4 option to save CP.pdb, but when I open it by gauss view, I see the following image:
#8 Multiwfn and wavefunction analysis » QTAIM topological graphs for SSH connection » 2025-01-28 09:19:23
- rezabma
- Replies: 8
Hi
I need topological graphs for BCP, RCP, and CCP from Bader theory (QTAIM). I know how can I save a text file for all CP properties. My connection is SSH, and I have only terminal of a Linux. How can I save the topological graph by a ssh connection?
Thanks
Reza
#9 Re: Multiwfn and wavefunction analysis » ODI » 2022-11-22 09:13:52
Dear Tian
Very thanks for your efforts. I will use SDI instead of ODI in my new paper. I have added two papers for citation: 1- J.Comp. Chem. for Multiwfn and 2-Acta Chimica Sinica 2011 for ODI. I will see the new version of the manual, and add another paper for citation if needed.
Thanks
Reza
#10 Re: Multiwfn and wavefunction analysis » ODI » 2022-11-20 08:38:54
Hi Tian
OK. Thank you for your fast reply.
I will start data production. I will cite Multiwfn and also the reference paper.
Best Regards
#11 Re: Multiwfn and wavefunction analysis » ODI » 2022-11-20 06:35:14
Dear Tian
Could you double-check the code, please?
For a cube with more density distribution, I get less ODI.
The integral over dr in ODI (dens) is the whole of the lattice, right?
I have uploaded the images of two cube files. the first is band number 307_alpha, and the second is band number 309_alpha.
For the first I get ODI=7.88 and for the second I get 26.78
for two images I set an iso-surface value of 0.001.
I can email these cubes if you need them.
Thanks
Reza
=========================================
307_alpha with ODI of 7.88:
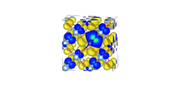
===============================================
309_alpha with ODI of 26.78:
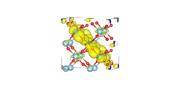
=====================================================
#12 Re: Multiwfn and wavefunction analysis » ODI » 2022-11-20 05:34:25
Dear Tian
Great!
Thanks for your efforts. I will check it.
With the Best Regards
Reza
#13 Re: Multiwfn and wavefunction analysis » ODI » 2022-11-17 07:59:40
Dear Tian,
I have emailed two cube files for two bands of doped TiO2.
#14 Re: Multiwfn and wavefunction analysis » ODI » 2022-11-15 15:57:26
Thanks, Tian,
I will calculate the density of some bands in the (cobalt and nitrogen) dual-doped Rutile phase by FHI-aims (some bands in the valence band and also the dopant band in the gap region). After emailing, I will inform you through this site.
#15 Multiwfn and wavefunction analysis » ODI » 2022-11-14 10:38:23
- rezabma
- Replies: 10
Hi
is it possible to use the orbital delocalization index for some bands in crystals (periodic cases)? I can generate cube files for the eigenstates (real or imaginary parts) of the bands, and also their density.
Please note that my code is FHI-aims, and the cube format is Gaussian cube format.
Thanks
Reza
#16 Multiwfn and wavefunction analysis » MultiWFN and Periodic DFT » 2022-01-08 05:32:49
- rezabma
- Replies: 1
Hi,
I know that you have tried to incorporate periodic calculations in MultiWFN. However, it has been restricted to CP2K code. Could you please add a module to the code provides availability of other pseudopotential/full potential LCAO solid DFT codes such as siesta/openmx or FHI-aims/Dmol3?
#17 Re: Multiwfn and wavefunction analysis » How invoke g16 in linux in parallel mode? » 2020-05-02 09:45:17
To be sure, let me check:
f-: how much the site is ready to donate its electron. means that how much the site is nucleophile.
f0: how much the site is ready for sharing electron and forming covalent bond.
f+: how much the site like to get more electrons. For example, by leaving H+ and getting minus charge.
So, O* atom in strong acids containing COO*H group should have high f+ and low f-.
Is it right?
#18 Re: Multiwfn and wavefunction analysis » How invoke g16 in linux in parallel mode? » 2020-05-02 05:19:38
Thanks.
I have more question about Fukui index. In the Multiwfn manual, there is an example for calculating f- f0 f+ for phenol. The data in manual shows that f- is larger for ortho and para sites. This is in line with experiments (especially in organic chemistry) which says that ortho and para sites in phenol have more electron density and ready for doing electrophilic attack (they are nucleophiles).
However, I see in some papers that interpret f- as the index that shows sites are more favorable to accept electrophilic attack (means they are positive sites and accept electrons,i.e., they are electrophiles)!!
The formula shows that f-=rho(N) - rho(N-1) which shows the molecules in these sites is ready to donate its electron. Please clarify this. It seems that in some papers (even Q1) it was misunderstood.
Thank You
#19 Re: Multiwfn and wavefunction analysis » How invoke g16 in linux in parallel mode? » 2020-04-30 05:47:24
Thanks. Solved.
Now I finished calculating three wfn files by running parallel job by invoking g16. I calculated quantitative indices (Number 2 in list). the program saved me a file named CDFT.txt.
I used B3LYP/6-31++G** for calculating WFNs. But in CDFT.txt I see following comment at the top of the file:
Note: the E(HOMO) of TCE used for evaluating nucleophilicity index is the value evaluated at B3LYP/6-31G* level
Well, I defined B3LYP/6-31++G** during all steps whenever it needed. I want to report the data in paper. How can force Multiwfn to use my level (B3LYP/6-31++G**) for above comment? Please note that I successfully defined my level in previous invoking steps, and I checked it in generated GJF files before running by G16.
Thanks
#20 Multiwfn and wavefunction analysis » How invoke g16 in linux in parallel mode? » 2020-04-29 11:28:41
- rezabma
- Replies: 7
Hi,
I run multiwfn in linux to obtain f-, f0, and f+ (Fukui functions). It needs that g16 invoked to calculate N-1.gjf, N+1.gjf, and N.gjf. I can generate three above files. I set g16 path by "/home/g16/g16". I have 2 problems:
1- in gjf files I see that multiwfn wants ti use 1 processor. How can I force it to use commands such as %nprocshared=44 %mem=200GB?
2-After generating three gjf file it could not invoke g16, and prints error:
Running: "N.gjf" "N.out"
sh: N.gjf: command not found
forrtl: No such file or directory
My gjf file is in /home/runs
My g16 id in /home/g16
My Multiwfn is in /home/Multiwfn
I run the multiwfn from my runs directory. I set g16 environment variables by "source /home/g16/bsd/g16.profile"
Thanks
#21 Re: Multiwfn and wavefunction analysis » ESP for periodic cube » 2020-03-02 06:18:00
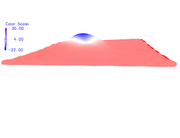
Very thanks. I hope that in the future Multiwfn could read and manipulate periodic jobs. My another question is that when I use density.cube and esp.cube (according to the above method), the summary analysis in Multiwfn says that positive surface area is 0.000 and positive average value is also NaN.
My charge data in FHI-aims show that in Al-doped graphene, Al has positive charge and carbons have near to zero but negative charges. In VMD, the color scale bar shows that Al has negative ESP, and C atoms have positive ESP. I have attached both figures. I think if this problem is solved, I could use the ESP figures in the paper.
Very thanks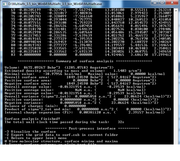
#22 Re: Multiwfn and wavefunction analysis » parallel job » 2020-03-01 17:10:49
Thanks. problem solved. Sorry for this carelessness.
#23 Re: Multiwfn and wavefunction analysis » ESP for periodic cube » 2020-03-01 16:16:58
Hi, Thanks for your efforts. I tested above method for my system (periodic Al-doped Graphene), and could generate the pdb file to enter in VMD. However, as you see in the attached file, the origin shifted. In FHI-aims, the origin for my system has been defined in:
cube origin 7.5089 4.3246 9.9289
cube edge 100 0.1500 -0.0868 -0.0012
cube edge 100 0.0002 0.1733 -0.0002
cube edge 100 0.0000 0.0000 0.2000
which shows the origin is x=7.5089, y=4.3246, and z=9.9289.
But seems that Multiwfn automatically choose (0,0,0) for the origin. I have two problems:
1- setting the origin in (7.5089, 4.3246, 9.9289)
2- how to remove layer in the z=20 of the cell (upper layer).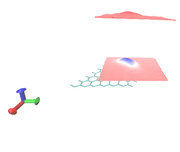
#24 Multiwfn and wavefunction analysis » parallel job » 2020-03-01 11:39:54
- rezabma
- Replies: 2
Hi,
On a 8-core system, I set 8 threads in setting file (on centos 7), but the maximum CPU% is 400 that means it uses only 4 core. How can set to use all 8 core?
#25 Re: Multiwfn and wavefunction analysis » ESP for periodic cube » 2020-03-01 07:44:58
OK. Thanks. I will test.
#26 Re: Multiwfn and wavefunction analysis » ESP for periodic cube » 2020-02-29 07:33:00
and this is a simple example (Acetylene cube) generated by FHI-aims and opened by GaussView. As you see there is not any number above it.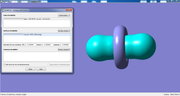
#27 Re: Multiwfn and wavefunction analysis » ESP for periodic cube » 2020-02-29 07:29:43
Please remember that the FHI-aims code is based on NAO (numeric atomic orbitals) similar to Dmol3.
#28 Re: Multiwfn and wavefunction analysis » ESP for periodic cube » 2020-02-29 07:28:26
Hi, Thanks for your rapid reply. I did following steps:
1- Optimization of the structure by FHI-aims
2- Generating cube file with "output cube hartree_potential" in FHI-aims (ESP cube)
3- reading the cube by Gaussview to check it is correct.
4- opening with multiwfn
5- going to 12
6- then to 0 (start analysis now)
7- getting NaN in "summary of surface analysis": see the picture.
I could upload the cube to test it if there is a place for uploading file (or email you)
Thanks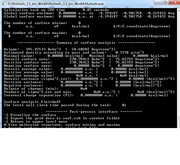
#29 Multiwfn and wavefunction analysis » ESP for periodic cube » 2020-02-23 18:46:11
- rezabma
- Replies: 22
Hi
I have generated a periodic cube file from FHI-aims code that contains ESP data (periodic graphene doped by Al atom). The cube is opened by multiwfn (and also GaussView), but displays NaN for minima and maxima ESP.
Any help.
Pages: 1