Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics: Active | Unanswered
#1 Re: Quantum Chemistry » Sigme- or Pi-hole characterization with Laplacian of Rho » 2025-03-23 03:14:31
Dear Tian,
Too many thanks for your kind attention and the highly valuable, informative, and professional suggestion.
Sincerely yours,
Saeed
#2 Re: Quantum Chemistry » Sigme- or Pi-hole characterization with Laplacian of Rho » 2025-03-22 19:52:10
Dear Tian,
Many thanks for your kind attention and, for guiding me with your highly valuable confirmation.
Please, also, let me ask one more question. In some cases, such as for TeHF compound in which the presence of a sigma-hole along the extension of the Te-F bond is expected, drawing isosurface of Laplacian of Rho (with isovalue= 0, namely the reactive surface) does not show a hole. On the other hand, this sigma-hole can be seen when isovalue is increased to 0.001 (a value slightly greater than zero). Could you please let me know why?
As another case, SnH3F could be mentioned. For this compound, the emergence of an expected hole along the Sn-F bond can never be satisfied using ANY value as isosurface for Laplacian of Rho. Such cases made me quite confused. Please let me know why some cases fail to present an expected sigma-hole over the Laplacian isosurface. I tried several times to attach "SnH3F.fch" file generated at M06-2X-D3(0)/def2-TZVPP level but, all tries failed! Using Multiwfn and VMD, I could not see the sigma-hole in this compound at any isovalue of Laplacian of Rho!
Sincerely,
Saeed
#3 Quantum Chemistry » Sigme- or Pi-hole characterization with Laplacian of Rho » 2025-03-21 18:39:05
- saeed_E
- Replies: 4
Dear Tian,
I hope you are doing well. Please, if possible, kindly let me ask you a question about Sigme- or Pi-hole characterization with the Laplacian of Rho.
Is it true to state that "a sigma-hole or a pi-hole is always characterized with the emergence of a hole on the isosurface of the Laplacian of Rho when an isovalue of zero is considered"?
In advance, thank you very much.
Sincerely,
Saeed
#4 Re: Quantum Chemistry » An unexplainable observation in the EDA » 2025-03-15 05:42:38
Dear Tian,
Many thanks for your highly valuable and informative comments.
Interestingly, as you have nicely recommended, the value of electrostatic interactions is quite consistent with the value of V_s,max calculated for heavy atoms in LAs.
Sincerely,
Saeed
#5 Quantum Chemistry » An unexplainable observation in the EDA » 2025-03-13 22:02:11
- saeed_E
- Replies: 2
Dear Tian,
Please, if possible, let me respectfully ask you a question regarding some unexplainable results in EDA I have recently encountered.
Please suppose 1,3-Aza Diene (A), including a sp2 hybridized nitrogen atom in position 2, interacts with some Lewis acids leading to inter-molecular tetrel-, pnictogen-, chalcogen-, and halogen-bound complexes (these LAs are SnH3F, SbH2F, TeHF, and FI all include a sigma-hole in the extension of heavy atom-F bond). These complexes participate in an Aza-Diles-Alder reaction toward a given dienophile (let's to be acetylene).
My energy decomposition energy analysis (EDA) indicates that electrostatic interaction is the main predominant player in stabilizing complexes considered. To explore the origin of the electrostatic interaction predominance, I calculated the atomic charge (Hirshfeld, NPA, VDD) on the heavy atoms of LAs. Unfortunately, computed atomic charges are not consistent with the values of electrostatic interactions. Indeed, while the positive charge on the heavy atoms of LAs decreases (becomes less positive) the electrostatic interaction between heavy atoms in LAs and nitrogen of Aza Diene increases (becomes more negative or stabilizing).
If possible, please let me know how you explain this strange and inconsistent observation.
In advance, please accept my highest gratitude for your kind attention to guiding me with your professional and golden comments and, please excuse me for bothering you.
Sincerely yours,
Saeed
#6 Re: Multiwfn and wavefunction analysis » Electron Density Surfaces For Individual Localised Orbitals » 2025-03-11 19:22:18
Thank you very much.
Saeed
#7 Re: Multiwfn and wavefunction analysis » Electron Density Surfaces For Individual Localised Orbitals » 2025-03-10 09:53:36
Dear Tian,
Many thanks for your much valuable comments.
If one wants to compare results of the gas phase and those of the solution, a quite same isovalue should be employed. In this sense, it seems isovalue= 0.001 a.u. to be suitable for both the gas and solution phase. Do you agree?
Sincerely,
Saeed
#8 Re: Multiwfn and wavefunction analysis » Electron Density Surfaces For Individual Localised Orbitals » 2025-03-09 20:22:18
Dear Tian,
In the above replies you indicated:
"the Bader's definition of molecular vdW surface (in gas phase) corresponds to isosurface of electron density with isovalue of 0.001 a.u.;.....". If possible, please let me know what is the suitable isovalue for the vdW electron density in the presence of a desirable solvent.
Sincerely yours,
Saeed
#9 Re: Multiwfn and wavefunction analysis » Drawing BCPs and Laplacian isosurface both at the same time? » 2025-03-05 11:42:37
Thank you very very much.
Best regards,
Saeed
#10 Re: Quantum Chemistry » Does M05-2X also need correction like M06-2X? » 2025-03-05 09:52:03
Dear Tian,
Too many thanks for your highly valuable and determining agreement and confirmation. In light of your highly valuable opinion, now I am ensured that my approach does not include any problem.
Sincerely,
Saeed
#11 Re: Multiwfn and wavefunction analysis » Drawing BCPs and Laplacian isosurface both at the same time? » 2025-03-05 09:46:48
Dear Tian,
Please accept my highest and deepest gratitude for the highly valuable time, energy, and patience you kindly spent to guide me in the best possible manner.
Following your instruction I could draw what I was looking for.
If possible, please let me ask a conceptual question. Considering my system I used "cub Laplacian 0.0" command in the VMD main windows so that the zero value is adjusted for both positive and negative parts of the Laplacian isosurface. As you can see in the presented picture, the selected BCP is located outside the zero isovalue of Laplacian. Consequently, it means the Laplacian at this BCP should be positive. Moreover, if a given BCP is located inside the zero isosurface of Laplacian (BCP is surrounded by Laplacian isosurface whose isovalue is zero), it means that Laplacian at that point should be negative. Please, if possible, let me know if my interpretations about the sign of Laplacian are reasonable based on how a BCP is located inside or outside of isosurface.
Sincerely yours,
Saeed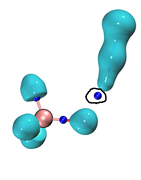
#12 Quantum Chemistry » Does M05-2X also need correction like M06-2X? » 2025-03-05 01:38:45
- saeed_E
- Replies: 2
Dear Tian,
It is well known that M06-2X functional has already been somewhat corrected during parametrization. So, to include dispersion corrections into this functional, one just needs to use "em=gd3". Please let me know has M05-2X also already been corrected to some extent during parametrization. If so, to include dispersion corrections, this functional also needs "em=gd3" keyword. Do you agree and confirm this statement?
In addition, I am computing solvation energy for a set of compounds at M05-2X/6-31G(d) level together with SMD solvation model as recommended by Truhlar and, as you also confirmed in one of your highly valuable blog articles. Please let me know if there are any problems regarding the accuracy of these calculations.
Sincerely yours,
Saeed
#13 Re: Multiwfn and wavefunction analysis » Drawing BCPs and Laplacian isosurface both at the same time? » 2025-03-04 09:41:59
Dear Tian,
Too many thanks for your so valuable guidance. But please let me state that I want to do visualization using a third party. Indeed, I want to generate corresponding "cub" files and, then, using "Cub" script in the VMD to reach very nice and high quality isosurface with BCPs. Could you please guide me to know how I can reach this purpose?
Sincerely,
Saeed
#14 Multiwfn and wavefunction analysis » Drawing BCPs and Laplacian isosurface both at the same time? » 2025-03-03 18:42:34
- saeed_E
- Replies: 6
Dear Tian,
Please let me ask a question regarding an issue I saw in an interesting article.
Please suppose one wants to draw BCPs together with the Laplacian isosurface exactly as presented in the attached picture.
As you can see, the Laplacian isosurface corresponding to isovalue 0.00 (namely reactive surface) and also BCPs are portrayed together. If possible, please let me know how one can reach such a picture using Multiwfn. Indeed, while I know how to draw Laplacian isosurface (5 Output and plot specific property within a spatial region and...), I do not know how to include BCPs at the same time.
Sincerely,
Saeed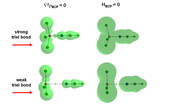
#15 Re: Quantum Chemistry » A question regarding SAPT analysis using def2 bases » 2025-03-01 15:02:22
Dear Tian,
Many thanks for your kind attention to prompt reply with highly valuable guidance.
So, given your nice comment, File 1 should be correct.
Moreover, I asked this question on the PSI4 forum but, unfortunately, and the same as other questions I did not receive any reply!
I also performed many searches on the net to find an appropriate response. Please be aware that the only valid file I could find regarding this question is "https://github.com/psi4/psi4/blob/maste … /input.dat". Interestingly, in quite agreement with your highly valuable recommendation, this file also uses "def2-SVP-ri" basis set as the orbital basis set is also "def2-SVP".
Once again, too many thanks.
Sincerely,
Saeed
#16 Quantum Chemistry » A question regarding SAPT analysis using def2 bases » 2025-03-01 09:59:19
- saeed_E
- Replies: 2
Dear Tian,
I hope you are doing well and, like always, kindly let me ask a question on my problem about SAPT analysis.
Please suppose one wants to perform SAPT analysis on a dimer, including heavy elements, for which using "def2" bases is mandatory. Let us take "def2-TZVPP" as the desirable basis set for such a give analysis.
It seems in the recent versions of PSI4 one should use "def2-universal-jkfit" instead of "def2-TZVPP-jkfit". The question is:
Is it also mandatory to employ "def2-universal-ri" instead of "def2-tzvpp-ri"? In other words, which of the below input files are reasonable and should be used (As I checked, the results are somewhat different:
File 1:
memory 55 gb
molecule {
0 1
C -1.56109100 -1.20061800 -0.00000400
C -0.17334400 -1.20786600 0.00002200
C 0.50607200 -0.00007600 0.00002300
C -0.17333700 1.20786200 0.00001700
C -1.56097000 1.20068700 -0.00000300
C -2.25745700 0.00000900 -0.00001500
H -2.09690700 -2.13988300 -0.00002400
H 0.37960100 -2.13607100 0.00000800
H 0.37980500 2.13594700 0.00001000
H -2.09684600 2.13992100 -0.00001000
H -3.33850200 0.00010300 -0.00001600
Cl 2.24080100 0.00000000 -0.00001200
--
2 1
Hg -0.77409920 -0.36639646 -1.52722807
units angstrom
no_reorient
symmetry c1
}
set {
basis def2-tzvpp
df_basis_scf def2-universal-jkfit
df_basis_sapt def2-tzvpp-ri
scf_type DF
guess sad
freeze_core True
}
set_num_threads(8)
energy('sapt2')
/////////////////////////////////////
File 2:
memory 55 gb
molecule {
0 1
C -1.56109100 -1.20061800 -0.00000400
C -0.17334400 -1.20786600 0.00002200
C 0.50607200 -0.00007600 0.00002300
C -0.17333700 1.20786200 0.00001700
C -1.56097000 1.20068700 -0.00000300
C -2.25745700 0.00000900 -0.00001500
H -2.09690700 -2.13988300 -0.00002400
H 0.37960100 -2.13607100 0.00000800
H 0.37980500 2.13594700 0.00001000
H -2.09684600 2.13992100 -0.00001000
H -3.33850200 0.00010300 -0.00001600
Cl 2.24080100 0.00000000 -0.00001200
--
2 1
Hg -0.77409920 -0.36639646 -1.52722807
units angstrom
no_reorient
symmetry c1
}
set {
basis def2-tzvpp
df_basis_scf def2-universal-jkfit
df_basis_sapt universal-ri
scf_type DF
guess sad
freeze_core True
}
set_num_threads(8)
energy('sapt2')
//////////////////////////////////////////////
In advance, your highly valuable guidance is very appreciated and, please excuse me for bothering you.
Sincerely yours,
Saeed
#17 Re: Quantum Chemistry » Computation of solvation energy » 2025-02-27 20:56:14
Dear Tian,
Your very valuable guidance is highly appreciated.
Sincerely yours,
Saeed
#18 Re: Quantum Chemistry » Computation of solvation energy » 2025-02-25 19:35:45
Dear Tian,
Thank you very very much for, like always, your highly valuable confirmation.
Please, if possible, let me request a reference regarding the reasonableness and valuability of the M06-2X/6-31G(d) level for the calculation of solvation energy.
Indeed, I need a reference (to be cited in the manuscript) in which the specific performance of the SMD solvation model as well as M06-2X/6-31G(d) level for the computation of solvation energy is addressed. I do not know whether the original SMD paper of Truhlar is sufficient or if there are some more appropriate references I am not aware of.
Sincerely,
Saeed
#19 Quantum Chemistry » Computation of solvation energy » 2025-02-25 11:52:22
- saeed_E
- Replies: 4
Dear Tian,
I hope you are doing well and all goes best with you.
If you kindly let me, I am respectfully going to ask a question regarding computation of salvation energy, Delta_E_solvation.
You know much better than me that Prof. "Truhlar" recommended using M06-2X/6-31G(d) computational level in conjunction with the SMD solvation model to compute solvation energy for chemical compounds in a very accurate manner whose results are very close to the experimental values.
Please suppose a compound including some elements for which 6-31G(d) basis set is not defined. In such cases, as you recommended in the long past (if I am not wrong), the def2-SV(P) could safely be used instead of 6-31G(d) basis hoping results are still satisfactory. Do you quite confirm the reasonableness of this statement?
In advance, your kind attention is highly appreciated.
Sincerely,
Saeed
#20 Re: Quantum Chemistry » An unknwn error in an Orca computation » 2025-02-20 06:51:38
Dear Tian,
Thank you very very much.
Interestingly, I completely understood your very valuable code, Molclus, and now I can work with this nice code perfectly.
It is also very surprising that the task is aborted if memory to be much more than is needed. Commonly, the low amount of memory encounters problem not the high amount!
Sincerely yours,
Saeed
#21 Re: Quantum Chemistry » An unknwn error in an Orca computation » 2025-02-20 01:48:44
Dear Tian,
Thank you very much for your prompt reply with informative comments.
I also have seen such scripts in the Orca 6 tutorials (https://www.faccts.de/docs/orca/6.0/tut … rapol.html) but, at least for me, they are not very clear. And, I think "Extrapolate" keyword is really so nice and practically useful. If possible, please let me share with you two issues: 1) Can I install both older and newer versions together on the same system? Indeed, while the older version worked best for some keywords, the newer version provides some very nice features such as "conformed search".
2) Regarding the problem mentioned, I performed exactly the same calculation but on a system including fewer atoms, and, the task was terminated normally. Given that I used 8 processors any of which with 6000MB of ram, it seems that 8*6000=48GB exceeded the total AVAILABLE ram and, the task has been aborted in the first calculation. Do you agree with me?
Sincerely,
Saeed
#22 Quantum Chemistry » An unknwn error in an Orca computation » 2025-02-19 22:33:13
- saeed_E
- Replies: 4
Dear Tian,
If possible, please let me state that I have frequently faced problems since upgrading my Orca 5.0.3 to 6.0.1; I have never encountered such problems with the older version.
Please let me ask a question regarding a new problem. The below Orca task was ran:
***********************************************************************************************************************
! ExtrapolateEP2(3/4,def2,MP2) tightSCF noautostart miniprint
%pal nprocs 8 end
%maxcore 6000
* xyz 0 1
C -3.00886523 -0.60110479 0.00000305
H -3.83275726 0.09558074 0.00003101
H -3.13625859 -1.67101162 -0.00011453
N -1.81764115 -0.14353739 0.00008421
H -0.98978097 -0.78273714 -0.00002296
O -1.48821106 1.09766974 0.00016013
C 1.46382272 -0.12319892 -0.00021950
O 0.71189603 -1.08884815 -0.00013887
C 2.95709267 -0.26671500 0.00015158
H 3.36553743 0.23708253 -0.87460156
H 3.36370189 0.22895946 0.88045529
H 3.23148130 -1.31574290 -0.00408829
O 1.05435101 1.11577850 -0.00024573
H 0.02497543 1.16194217 -0.00006346
*
$new_job
! ExtrapolateEP2(3/4,def2,MP2) tightSCF noautostart miniprint Pmodel
%pal nprocs 8 end
%maxcore 6000
* xyz 0 1
C -3.00886523 -0.60110479 0.00000305
H -3.83275726 0.09558074 0.00003101
H -3.13625859 -1.67101162 -0.00011453
N -1.81764115 -0.14353739 0.00008421
H -0.98978097 -0.78273714 -0.00002296
O -1.48821106 1.09766974 0.00016013
C : 1.46382272 -0.12319892 -0.00021950
O : 0.71189603 -1.08884815 -0.00013887
C : 2.95709267 -0.26671500 0.00015158
H : 3.36553743 0.23708253 -0.87460156
H : 3.36370189 0.22895946 0.88045529
H : 3.23148130 -1.31574290 -0.00408829
O : 1.05435101 1.11577850 -0.00024573
H : 0.02497543 1.16194217 -0.00006346
*
$new_job
! ExtrapolateEP2(3/4,def2,MP2) tightSCF noautostart miniprint Pmodel
%pal nprocs 8 end
%maxcore 6000
* xyz 0 1
C : -3.00886523 -0.60110479 0.00000305
H : -3.83275726 0.09558074 0.00003101
H : -3.13625859 -1.67101162 -0.00011453
N : -1.81764115 -0.14353739 0.00008421
H : -0.98978097 -0.78273714 -0.00002296
O : -1.48821106 1.09766974 0.00016013
C 1.46382272 -0.12319892 -0.00021950
O 0.71189603 -1.08884815 -0.00013887
C 2.95709267 -0.26671500 0.00015158
H 3.36553743 0.23708253 -0.87460156
H 3.36370189 0.22895946 0.88045529
H 3.23148130 -1.31574290 -0.00408829
O 1.05435101 1.11577850 -0.00024573
H 0.02497543 1.16194217 -0.00006346
*
$new_job
! ExtrapolateEP2(3/4,def2,MP2) tightSCF noautostart miniprint Pmodel
%pal nprocs 8 end
%maxcore 6000
* xyz 0 1
C -3.00886523 -0.60110479 0.00000305
H -3.83275726 0.09558074 0.00003101
H -3.13625859 -1.67101162 -0.00011453
N -1.81764115 -0.14353739 0.00008421
H -0.98978097 -0.78273714 -0.00002296
O -1.48821106 1.09766974 0.00016013
*
$new_job
! ExtrapolateEP2(3/4,def2,MP2) tightSCF noautostart miniprint Pmodel
%pal nprocs 8 end
%maxcore 6000
* xyz 0 1
C 1.46382272 -0.12319892 -0.00021950
O 0.71189603 -1.08884815 -0.00013887
C 2.95709267 -0.26671500 0.00015158
H 3.36553743 0.23708253 -0.87460156
H 3.36370189 0.22895946 0.88045529
H 3.23148130 -1.31574290 -0.00408829
O 1.05435101 1.11577850 -0.00024573
H 0.02497543 1.16194217 -0.00006346
*
******************************************************************************************************************************************************************
But after a long time, the task was terminated and the below message was displayed on the screen:
--------------------------------------------------------------------------
Primary job terminated normally, but 1 process returned
a non-zero exit code. Per user-direction, the job has been aborted.
--------------------------------------------------------------------------
--------------------------------------------------------------------------
mpirun noticed that process rank 2 with PID 0 on node saeed exited on signal 9 (Killed).
--------------------------------------------------------------------------
[file orca_tools/qcmsg.cpp, line 394]:
.... aborting the run
***************************************************************************************************************************************************************
Moreover, at the bottom of the output file, one can see:
Maximum memory used throughout the entire LEANSCF-calculation: 57.2 MB
************************************************************
* Program running with 8 parallel MPI-processes *
* working on a common directory *
************************************************************
------------------------------------------------------------------------------
ORCA MP2
------------------------------------------------------------------------------
Freezing NCore=14 chemical core electrons
----------
MP2 ENERGY (disk based algorithm)
----------
Dimension of the basis ... 609
Memory devoted to MP2 ... 6000 MB
Data format for buffers ... DOUBLE
Compression type for matrix containers ... UNCOMPRESSED
-------------------------
SHARK HALF TRANSFORMATION (Exchange order)
-------------------------
Number of basis functions ... 609
Number of operators ... 1
Operator 0: 7- 27 (p-index in (p*|q*))
Operator 0: 7- 27 (q-index in (p*|q*)
Integral generator used ... SHARK
Contraction scheme used ... SEGMENTED CONTRACTION
MaxCore in resort ... 6000 MB
Half transformed integrals for op= 0 ... R1_S.SHARK_MNPQ0.tmp
Resorted half transformed integrals ... R1_S.KAO_aa.tmp
ORCA finished by error termination in MP2
Calling Command: mpirun -np 8 /home/saeed/orca-6.0.1/orca_mp2_mpi R1_S.mp2inp.tmp R1_S
[file orca_tools/qcmsg.cpp, line 394]:
.... aborting the run
Please, if possible and like always, make me benefit of your highest kindness and let me know how I can resolve this problem.
In advance, many thanks for your valuable time and energy and, please excuse me for bothering you.
Sincerely,
Saeed
#23 Re: Multiwfn and wavefunction analysis » There is a problem when ELF colored-filled map is generated » 2025-02-18 07:58:09
Dear Tian,
Thank you very much; the problem was resolved in the light of your so valuable guidance.
Indeed, before choosing option 1 (regarding plotting in the XY plane), I should select option 0 (Set extension distance for plane type 1~5, current:) and, slightly increase the current number.
Sincerely,
Saeed
#24 Multiwfn and wavefunction analysis » There is a problem when ELF colored-filled map is generated » 2025-02-18 07:29:49
- saeed_E
- Replies: 2
Dear Tian,
I have a structure for which the ELF colored-filled mao is going to be generated (4--->9--->1--->1 (XY plane)). Indeed, the generated picture is larger than that can be fixed between X and Y axes. In other words, some parts of figure are covered by the X and Y axes and, I need to extend these axes so that all parts of figure are evidently seen. If possible, please let me know how I can resolve this problem.
Sincerely,
Saeed
#25 Re: Quantum Chemistry » Which basis set do you recommend for Transition metals-included specie » 2025-02-14 06:11:35
Dear Tian,
OK, and many many thanks.
Sincerely,
Saeed
#26 Re: Quantum Chemistry » Which basis set do you recommend for Transition metals-included specie » 2025-02-13 23:05:33
Dear Tian,
Thank you very much for your kind attention to guide me, like always, in the best possible manner.
Could you please let me know what (-f) means in this basis set and how such a basis set can be reached (or constructed)? I did not find this basis in the "BSE.org". I have not ever used such bases.
In advance, please excuse me for taking your valuable time.
Sincerely yours,
Saeed
#27 Quantum Chemistry » Which basis set do you recommend for Transition metals-included specie » 2025-02-13 10:45:42
- saeed_E
- Replies: 4
Dear Tian,
Good day! I hope you are doing well.
I remember you recommended TPSSh functional with D3(BJ) correction for geometry optimization and energetics study of transitional metal-included complexes. Please let me know which basis set you recommend in conjunction with this functional to reach the mentioned purpose. I have frequently seen, whenever possible, researchers use aug-cc-pVTZ basis set. Do you agree with this basis set? It seems DFT functionals display the best performance in conjunction with def2-series bases while post-HF ones are better work with cc-bases.
Sincerely,
Saeed
#28 Re: Quantum Chemistry » How to used GaussView-like formats in VMD » 2025-01-26 09:55:37
Dear Tian,
Thank you very much.
All was done correctly!
Sincerely,
Saeed
#29 Quantum Chemistry » How to used GaussView-like formats in VMD » 2025-01-26 03:11:27
- saeed_E
- Replies: 2
Dear Tian,
One of your nice articles is associated to perform some initial preparations to force VMD acts as GaussView formats. I remember that there is a nice article in Chinese for this purpose. Could you please let me know the exact address of this article?
Sincerely,
Saeed
#30 Re: Quantum Chemistry » VMD version » 2025-01-25 10:21:27
Dear Tian,
Thank you very much for your kind attention, valuable time, and highly valuable recommendation.
As far as I performed many tests, there were not any problems regarding visualization and performance of 1.9.1 version. Please, if possible, let me know if there is any specific problem in this version which I am not aware of that. Otherwise, due to slightly difficult removing and re-installing software in Ubuntu, I prefer to keep hold this version.
Sincerely,
Saeed