Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
#1Multiwfn and wavefunction analysis»Intel MKL error: Parameter 8 was incorrect on entry to DGEMM»2022-04-20 05:57:05
- julieborah66
- Replies: 1
-
Dear Sir,
I have a molecule ( consisting of 104 atoms).
I want to calculate the RMSE and RRMSE ( ESP error). I have selected a file containing charges ( RESP Charges) the .chg file and loaded the option 7 Population analysis and atomic charges
Then I went to the 13 Merz-Kollmann (MK) ESP fitting atomic charge.
Again selected the .chg file and then I set the number and scale factors of layers of MK fitting points (1.67 to 2.20)
I selected the 49th atom
Then this is the error I got
Intel MKL error: Parameter 8 was incorrect on entry to DGEMM and then the Multiwfn software stopped abruptly.
A picture has been attached for reference (P 2. png). With the rest of the atoms, it was giving me values but with two atoms I am getting this error! Also, I would like to add that I have done similar RMSE and RRMSE analyses of CM5 and PCM-CM5 atomic charges, it was working fine!
I did the same thing on my Desktop ( to check if this is a core issue) and then I got NAN issue ( the reference photo: 49 atom problem.png)
Please, help me to sort out the problem!
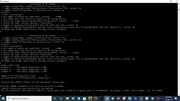

#2Multiwfn and wavefunction analysis»Calculate the dipole moment using different partial charges»2020-09-17 07:20:06
- julieborah66
- Replies: 1
-
Dear Sir,
Is there a provision in Multiwfn to calculate the dipole moment using partial charge( user specified) in Multiwfn?
#3Re:Multiwfn and wavefunction analysis»Control Grid spacing,Extension distance (in 3directions) in ESP»2020-03-01 17:35:16
Thanks, Sir, I would like to ask one more thing, does the second last column of ESPfitpt.pqr record the ESP values of the point? For example a pqr file
HETATM 1 O MOL A 1 16.939 -15.091 -0.001 -9.94175506 0.100O
HETATM 2 O MOL A 1 17.235 -14.795 -0.001 -9.19897403 0.100O
#4Re:Multiwfn and wavefunction analysis»Control Grid spacing,Extension distance (in 3directions) in ESP»2020-03-01 05:11:44
Thank you, Sir, for your prompt reply. So, is there a way in Multiwfn to get RMSE and RRMSE of a certain atom using a set of charges specified by the user but the user has control over the grid points in ESP like spacing and extension distance? Actually I am trying to implement a procedure as given in the paper--- Kramer, C., Spinn, A., and Liedl, K. R. “Charge anisotropy: Where atomic multipoles matter most”. Journal of Chemical Theory and Computation, 10(10), pp. 4488–4496 (2014)
#5Re:Multiwfn and wavefunction analysis»Control Grid spacing,Extension distance (in 3directions) in ESP»2020-02-29 04:21:29
Thank you, Sir, I am doing example 4.7.8 of the manual, Examine electrostatic potential reproducibility of atomic charges, is there a way to control the grid spacing in RMSE comparison of a particular atom? and what is the default grid spacing if not set? I need a 5 Angstrom extension distance around the molecule. So, I changed in Aug 3D (converting Angstrom to Bohr 9.44 Bohr) and I want 0.3 Angstrom grid spacing.
#6Multiwfn and wavefunction analysis»Control Grid spacing,Extension distance (in 3directions) in ESP»2020-02-28 16:43:35
- julieborah66
- Replies: 7
-
Dear Sir,
I have a small query, Is it possible to have a control over the grid points and Extension distance ( in x, y and z directions) from the molecule in calculation of ESP? Will change in the settings.ini in Aug3D would serve the purpose for the extension distance? Do we have an input variable for grid spacing too in settings.ini?
#7Re:Quantum Chemistry»Total sum of charge of CM5»2019-03-08 20:13:55
Thank you Sir for helping me out. I my full of gratitude.
#8Re:Quantum Chemistry»Total sum of charge of CM5»2019-03-08 06:48:16
Sir, apologies for not making it understandable, by "octamer" I mean "two monomers "of the polymer used for calculation and by "long molecule" I mean "25 monomers" of the polymer used. I know RESP fitting is better than CM5, but for comparison of charges I do have to consider all the charges.
I took an octamer for calculation, since it is the best representation of the polymer by some experimental results. Now, the problem that arises is getting a neutral condition, So, I made some small adjustments to the charges obtained by DFT in the fourth decimal of the charges of atoms to make it a neutral for MD purpose.
Is there any better way than this to obtain CM5 charges for the polymer (long chain)?
#9Quantum Chemistry»Total sum of charge of CM5»2019-03-06 11:33:04
- julieborah66
- Replies: 4
-
Hello Sir,
I am using an octamer (consist of 104 atoms) that has a first, middle and end residue. The CM5 charges are being generated by Multiwfn and total of the CM5 charges is zero,now I want to apply the charges to a long molecule (1162 atoms) and want to do a molecular dynamics simulation, then I use the charges for front residue for octamer to long molecule, middle residue for octamer to long molecule and so on, and now obviously the charges of the long molecule isn't a zero (for floating numbers), which is a pre-requisite for doing molecular dynamics simulation (the total charge of the molecule should be neutral), So, if i make small changes in the fourth decimal of the charge file in md, and get it a zero, am I proceeding in the right way? ( I did change the fourth decimal because playing up with parameters isn't allowed) .
#10Re:Multiwfn and wavefunction analysis»Low quality grid points»2019-03-06 05:04:04
Okay Sir, Thank you for helping me out. I will look at the distribution of points in VMD.
#12Re:Multiwfn and wavefunction analysis»Low quality grid points»2019-03-05 18:51:48
Thanks Sir for your prompt reply.
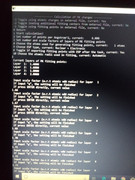
Here are my details, I want to calculate the RMSE of the .chg file with Quantum ESP using MK fitting mode
There is an option of selecting the MK layers in the menu, I want to select all the points lying in between 1.66 of the vdw radius to 2.20 of the vdw radius of a particular atom. I am giving the layers option as 1.66,1.67, 1.68...till 2.20, so 55 layers. Then it yields a statement that 11020 points are being used. I am attaching some pics of the screen where commands are mentioned.
So, I would like to know from you if I am doing it the correct way?
If this method is wrong is there any way to do this of getting RMSE of a specific atom within a particular range of vdw radius specified by user, using any fitting method in Multiwfn?
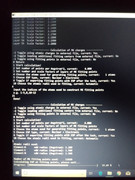
#13Multiwfn and wavefunction analysis»Low quality grid points»2019-03-05 10:25:55
- julieborah66
- Replies: 5
-
Sir,
For calculating the ESP of an octamer (that contains 104 atoms) the total number of grid points used for calculating the ESP of an atom is 10,000 grid points. Is this a reasonable value of grid points? Since, increasing it would cause computational longer time and also to be noted that my system is bigger.
#14Re:Multiwfn and wavefunction analysis»Calculation of solubility parameter from ESP»2018-12-15 19:19:15
Thank you Sir for your answer. I will surely do the protocol as mentioned.
#15Re:Multiwfn and wavefunction analysis»Calculation of solubility parameter from ESP»2018-12-15 09:55:46
I want to check if the point charges are used in the forcefield parameters will they be able to predict the solubility parameter (calculated by ESP) near to experimental values.
#16Re:Multiwfn and wavefunction analysis»Calculation of solubility parameter from ESP»2018-12-14 11:27:59
Is there any way out by which we can calculate the electrostatic potential map generated by the atomic charges which could assist to find out the solubility parameter?
#17Multiwfn and wavefunction analysis»Calculation of solubility parameter from ESP»2018-12-14 08:51:36
- julieborah66
- Replies: 6
-
I want to calculate the solubility factor from the ESP map as stated in (J. Mol. Struct. (THEOCHEM), 307, 55), but by taking the .chg file from a set of atomic charges.
Is it possible to do in Multiwfn, using the following procedure.
1. A .chg file loaded of the molecule
2. and going to the main function 12, Quantitative analysis of molecular surface
3. going to 0 Start the analysis.
4. Getting the product of total esp variance and degree of charge balance
5. Calculating the solubility parameter.
#18Quantum Chemistry»Electrostatic potential in a PCM model»2018-12-10 12:53:23
- julieborah66
- Replies: 1
-
Sir, the wave function file is affected by the PCM model, then in the calculation of the RMSE of atomic point charges with the quantum one does it accounts for the dielectric constant for example 80, since the total molecular electrostatic potential is a function of distance, atomic charge and a integral of charge density and it does not contain the dielectric constant.
So, if I want to compare the electrostatic potential of point charges with the Quantum one in a particular solvent, and since I am supplying a wave function with the value of dielectric constant. Can I take that the RMSE values calculated by multiwfn is the what I want, or for a answer I want do I have to divide the RMSE with the value of dielectric constant of solvent?
Any suggestion or comments is highly appreciated.
#19Multiwfn and wavefunction analysis»Queries to .chg file»2018-11-02 07:13:25
- julieborah66
- Replies: 1
-
Hello everyone,
I have a question on the .chg file generated by Multiwfn. Sir, I made a calculation on orca using the polarized continuum model using a dielectric constant of 80(water). I got the output converted to .wfn file. Now, when I calculate the CM5 or any other charges, using the .wfn file generated, does the .chg file take into consideration the dielectric constant value of water?
#20Re:Multiwfn and wavefunction analysis»Default grid spacing in Multiwfn»2018-10-22 10:18:42
Sir,
When the RMSE appears on the screen, what is it's unit?Is it in a.u. as mentioned in page 277
#21Re:Multiwfn and wavefunction analysis»Default grid spacing in Multiwfn»2018-10-21 03:45:30
Thank you Sir for correcting me. I will upload the C2H2.wfn file. However, I was wondering if there is a way out to specify the Merz-Kollman grid spacing points in calculation of the electrostatic potential. Any suggestion or comment is highly appreciated.
#22Re:Multiwfn and wavefunction analysis»Default grid spacing in Multiwfn»2018-10-20 11:42:26
Sorry Sir for the incomplete msg
I wanted to use my own specified grid setting and then calculate the RMSE of the molecule
For this I used the C2H2.wfn file. These are the steps I followed
1. Went to main function 5 (Output and plot specific property within a spatial region)
2. Then went available real space function option 12( Total electrostatic potential
3. Seting up grid by option 4 (grid spacing)
4. Inputting the grid spacing in bohr unit e.g 0.56 in all directions
5. Exporting the data via option 2 (a cube file generated)
6. I did some additional settings in order to change the extension distance of the molecule went to settings.ini and changed the Aug3D
7. Booted Multiwfn again
8. I had chosen the cube file generated before (step 5 I mentioned)( I wanted to have the grid setting as I had set so I uploaded the cube file)
9. Went to main function 7( Population analysis)
10. Then in Population analysis I opted for 13 ( MK mode)
11. Then I gave -3 as I wanted to compare the atomic charges from CM5 ( already produced stored in .chg file)
12. Gave the path to the file. Opted for option 3 to set the layers since I want the grid points lying within a particular range from 1.66 to 2.2 of vdw radius of a specified atom. I gave layer one 1.66 and layer two 2.2 so that only those grid points are considered lying in the range while calculating esp and others are not considered.
13. I then opted for option 4 choosing atoms. I gave 1 as input if I want to check the esp of 1st atom
14. Then selected 1 as option for starting calculation.
15. It gave me output
Sum of charges -0.000036
RMSE 2.29. RRMSE 1.000963
So by this can I take that the error in producing ESP of the carbon atom when measured with quantum esp is 2.29.
I am new to DFT and learning it so I am having problems
#23Re:Multiwfn and wavefunction analysis»Default grid spacing in Multiwfn»2018-10-20 06:00:25
Sir,
The procedure that I used to change the grid setting for my input file is as follows. Is it the correct way?
1. Loading the wfn file
2. Going to mode 5 setting the grid
3. Calculating the esp and changing the Aug3D for grid extension to 5 angstron beyond the molecule.
4. Changing the grid spacing in mode 6
5. Exporting the output to the cube file
6. Booting up Multiwfn again and loading the cube file with the grid setting
7. Calculating the atomic charge RMSE by entering the MK module
8. Changing the layer scale factor according to my specifications since I want only to consider those grid points lying in between a specified range.
The output is generated. So, can I assume I am doing correctly?
#24Multiwfn and wavefunction analysis»Default grid spacing in Multiwfn»2018-10-20 05:06:25
- julieborah66
- Replies: 9
-
Sir,
What is the default grid spacing in Multiwfn? Do we have an option in the settings.ini to change the value without going to the Mode 4 of the setup grid as we the option of changing the extension distance in settings.ini in Aug3D?
#25Re:Multiwfn and wavefunction analysis»Esp grid points»2018-10-14 03:53:50
Okay Sir, thankyou Sir for updating
#26Re:Multiwfn and wavefunction analysis»Esp grid points»2018-10-10 16:16:21
Thank you Sir for being so prompt in replying to queries. I do need an option where I can calculate the RMSE error of a specific atom in a given molecule, if I upload the chg file. It will be so useful if such an option will be available in Multiwfn
#27Re:Multiwfn and wavefunction analysis»Esp grid points»2018-10-10 12:08:57
Sir,
I uploaded a file containing CM5 charges. The phenol.wfn file was taken from the example section and then the CM5 charges were calculated. I uploaded the .chg file in the CHELP module I got RMSE error 0. I also did it with MK scheme. I got RMSE. Why is that so? Is there something wrong in my procedure. I am attaching a screenshot of the results
#28Re:Multiwfn and wavefunction analysis»Esp grid points»2018-10-10 11:43:50
Hello Sir,
Apologies to disturb you once again, is there any way out in Multiwfn to calculate the RMSE error around a specific atom?
#29Re:Multiwfn and wavefunction analysis»Esp grid points»2018-10-08 08:18:02
Hello Sir,
Thanks for your reply, Actually, I am referring to a paper " Charge Anisotropy: Where Atomic Multipoles Matter Most: Christian Kramer, Alexander Spinn, and Klaus R. Liedl, Journal of Chemical Theory and Computation, 2014, 10, 4488−4496" in the page 4490 of statistical analysis they mention a line " Each grid point is assigned to the nearest atom for the analysis " (grid points at a distance of 1.66−2.2 times the vdW radius)
How can I assign in my case if I want to calculate the RMSE of the ESP between the quantum and the CM5 charges. I want not using any fitting CHELP or RESP. It's just that I want to calculate to error metrics.
#30Multiwfn and wavefunction analysis»Esp grid points»2018-09-27 14:04:23
- julieborah66
- Replies: 11
-
Sir,
I generated esp (in grids) for my molecule of atomic charges and got it into a text file espfit.txt?
Is there a way to know which atom is assigned which grid points, since the text file contains x,y,z coordinate of the grid points and fourth column has esp but how can I know the atom numbers?