Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
Pages:1
#1Multiwfn and wavefunction analysis»sobEDA with Orca»2023-12-01 12:50:51
- michaelp
- Replies: 4
-
Dear Tian,
MultiWFN gets more and more useful. I find the sobEDA analysis quite interesting. I also appreciate your efforts for producing a very detailed description and even scripts. These scripts are meant to work with gaussian. Unfortunately I do not have access to gaussian. I was therefore wondering if it would be possible for you to provide a script that uses orca?
Kind regards,
Michael
#2Re:Multiwfn and wavefunction analysis»Visualisation of atomic quadrupole moments»2022-04-20 22:45:00
Dear Tian,
thank you very much again.
Best wishes,
Michael
#3Re:Multiwfn and wavefunction analysis»Visualisation of atomic quadrupole moments»2022-04-20 10:09:06
Dear Tian,
May I add a quick question?
Using the latest linux version of MultiWFN I have problems with the atom_moment.txt file generated in AIM basins. The file is empty. The procedure I use is the following:
Loading the orca molden file
17,1,1,4,8
then answering "y"
This produces two files "multipole.txt" and "atom_moment.txt". The first one looks like the file generated from fuzzy atom analysis, but the second file is empty. Could you point me to my error?
Thanks,
Michael
#4Re:Multiwfn and wavefunction analysis»Visualisation of atomic quadrupole moments»2022-04-20 09:48:11
Dear Tian,
Thank you very much for the implementation. I already tried it out on a few examples and it works flawlessly. I am grateful to you for adding this functionality so quickly to MultiWFN.
Best wishes,
Michael
#5Re:Multiwfn and wavefunction analysis»Visualisation of atomic quadrupole moments»2022-04-08 10:07:37
Dear Tian,
Thank you for your reply. This would indeed be very valuable!
Kind regards,
Michael
#6Re:Multiwfn and wavefunction analysis»Visualisation of atomic quadrupole moments»2022-04-07 17:51:12
If such a visualisation is not feasible, then calculating the eigenvectors of the Q tensor and exporting these as three Xe atoms would be great. With the help of this the form of the tensor (prolate vs. oblate) could be discerned.
Thank you in advance,
Michael
#7Re:Multiwfn and wavefunction analysis»Installing Multwfn on Mac M1 Monterey 12.3»2022-04-07 15:06:57
Hello,
I am both a Mac user and a MultiWFN user. In my opinion it is easiest, to run MultiWFN remotely on a linux machine. MultiWFN has great capabilities to produce gaussian cube files which can then be visualised by e.g. vmd on your Mac. All relevant data produced by MultiWFN can be written to files and then analysed on your Mac.
In principle you could try to run MultiWFN using wine or on a virtual machine. I have not tried wine, but a virtual machine would not work as the M1 has architecture that is not supported by MultiWFN. You could try to compile it on a virtual machine, but that would be quite an undertaking. I am quite sure, the remote machine option is the easiest way to use MultiWFN for you.
Kind regards,
Michael
#8Multiwfn and wavefunction analysis»Visualisation of atomic quadrupole moments»2022-04-06 14:47:29
- michaelp
- Replies: 9
-
Hello Tian,
thanks to your efforts it is possible to calculate atomic multipole moments based on the fuzzy atom approach. I was wondering, if there is an easy way to visualise the results. In principle if one looks at the ration of Q_xx/Q_zz one can plot a quadrupole moment distribution. For Q_xx/Q_zz = 0.5, this would look something like the attached figure.
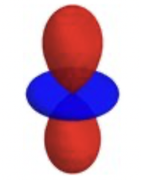
In the extreme of Q_xx being larger than Q_zz the graph would be rotated by 90° and the colours switched. Visualisation would be fairly straightforward, if the atom of interest was in the centre and the molecule suitably aligned... Alas that is almost never the case. Could you add a function, whereby one could create an isosurface of the quadrupole moment distribution of an atom in a molecule? I think this would be extremely helpful.
Best wishes,
Michael
#9Re:Quantum Chemistry»imposing a point group symmetry»2021-08-17 15:36:06
Hello,
the vmd program will do that for you as well in a very user friendly graphical environment. You will find the feature from the vmd main window under Extensions -> Analysis -> Symmetry Tool.
Regards,
Michael
#10Re:Multiwfn and wavefunction analysis»Reduce cost of ESP analysis on molecular surface for ORCA users»2021-01-19 08:00:40
Thank you for this post. It is indeed a much faster way to get the MEP. I have a problem though with the grid definition. I would like to visualise the ELF and colour-code it with the MEP. In order to do this I generate an ELF cube and I copied the spacing of the grid points from that calculation. I then follow your scheme, but I change the spacing to exactly the same value as used for the ELF cube. Still, the final MEP cube has different dimensions and can therefore not be used to colour code the ELF cube. Is there any way to lift this problem?
Kind regards,
Michael
#11Multiwfn and wavefunction analysis»Natural Orbitals of Excited States»2020-12-17 12:22:26
- michaelp
- Replies: 1
-
Hello,
I find the possibility to generate natural orbitals of specific excited states extremely helpful. I use this option regularly in combination with orca outputs. MultiWFN lets me choose between singlet and triplet excitations which is very convenient. I was wondering whether it would be possible to generate such information also in the case of a spin-orbit coupled calculation. I realise that it would be tricky as we are talking about a two-component wave function in principle. But it would already be helpful to get the natural orbitals as a weighted sum of the relevant singlet and triplet contributions to the desired SO-coupled state. Would it be feasible to integrate such a feature in MultiWFN?
Best wishes,
Michael
#12Multiwfn and wavefunction analysis»IQA analysis?»2020-07-17 09:38:48
- michaelp
- Replies: 1
-
Hello,
I was wondering if you are planning to include the method of interacting quantum atoms. It seems to be very useful to get quantitative data on e.g. covalency of a bond.
Best wishes,
Michael
#13Re:Multiwfn and wavefunction analysis»MacOS bin for MultiWFN 3.6 (dev)»2019-04-05 13:59:52
Dear Tian,
Thank you for your quick response. I am testing the Mac version now. So far everything checks out. I will continue to check against the Linux version.
Kind regards,
Michael
#14Multiwfn and wavefunction analysis»MacOS bin for MultiWFN 3.6 (dev)»2019-04-04 07:10:41
- michaelp
- Replies: 2
-
Hello,
would it be possible for you to compile the latest 3.6 dev version of MultiWFN for MacOS? I would like to use the new features, but I have no access to the intel fortran compiler for Mac!
Kind regards,
Michael Patzschke
#15Re:Multiwfn and wavefunction analysis»Basin analysis of density differences»2018-06-07 15:31:00
Sorry for posting a question I could answer myself. It is already possible!
Thank you
#16Multiwfn and wavefunction analysis»Basin analysis of density differences»2018-06-07 14:01:02
- michaelp
- Replies: 2
-
Hello,
It is very convenient to produce density difference plots with MultiWFN. When doing so, I noticed that it would be even more useful if it were possible to make a basin analysis of the density differences and then integrate the basins. Putting numbers to density differences would make them much more convincing. Is there already a way to do such an analysis in MultiWFN that escaped my attention? If not would it be possible to include such a feature in upcoming versions?
Kinds regards,
Michael
#17Multiwfn and wavefunction analysis»Why not NBO?»2018-03-19 13:48:41
- michaelp
- Replies: 1
-
Dear Tian,
MultiWFN is user friendly and full of options. It is almost a complete package for density and wave-function analysis. Thank you for the work and effort you put into this code. My question is, would it be possible to also include the NBO analysis? There is no obvious reason, why this should not be included and would make the code even more useful.
Best wishes,
Michael
#18Re:Multiwfn and wavefunction analysis»Compiling MultiWFN dev. version 3.5»2018-03-19 13:44:17
Dear Tian,
that did the trick! Thank you very much.
Best wishes,
Michael
#19Multiwfn and wavefunction analysis»Compiling MultiWFN dev. version 3.5»2018-02-27 11:48:46
- michaelp
- Replies: 2
-
Hello all,
I was trying to compile the development version of MultiWFN under linux. But the compilation fails. I am trying to compile the noGUI version and I have tried different versions of ifort. All versions (14-18) crash with the following error message:
/tmp/ifort2kAtx2.o: In function `MAIN__':
wfn.f90:(.text+0x742): undefined reference to `selfilegui_'
wfn.f90:(.text+0x4dd9): undefined reference to `igm_'
wfn.f90:(.text+0x4dec): undefined reference to `rdg_md_'
wfn.f90:(.text+0x4e10): undefined reference to `orbloc_'
make: *** [default] Error 1
Any ideas how to resolve the issue?
Thank you,
Michael
#20Re:Multiwfn and wavefunction analysis»NCP localisation for actinide compounds»2018-02-07 08:54:26
Dear Tian,
thank you for your fast reply! Loosening the convergence criteria did the trick. I tried to tighten the convergence criteria - that was the wrong way to go obviously. It finally worked with gradient norm convergence set to 1.0e-04 and displacement convergence at 1.0e-05.
Best wishes,
Michael
#21Multiwfn and wavefunction analysis»NCP localisation for actinide compounds»2018-02-06 10:00:55
- michaelp
- Replies: 2
-
Hello,
I have been using MultiWFN for teaching and research and I am very happy with the program. I recently stumbled upon a problem though. I have been calculating simple uranyl complexes with orca (all electron ZORA or DKH, trying also different DFT functionals) and in each case MultiWFN could not find the NCP of the uranium atom. I used a molden file generated by orca which I translated into a .wfn file by means of the molden2aim code. I also tried using the latest version of MutliWFN with a molden file generated by orca directly, no luck there either. I Would appreciate help in the matter.
Best wishes,
Michael
Pages:1