Spin multiplicity is not directly shown in spectrum, it is only indirectly reflected in the simulated spectrum (spin multiplicity affects position of NMR chemical shifts).
okok
do you know why Gaussian 16 doesn't show me the spin multiplicity in simulated spectra ?
i didn't find anything in literature.
Thanks
"0 1" in line 8 of your gjf file indicates net charge is 0, spin multiplicity is 1.
By the way, "=noraman" is completely redundant. For DFT, Gaussian doesn't calculate Raman activity by default.
How can i define it the spin multiplicity in Gaussian Input file?
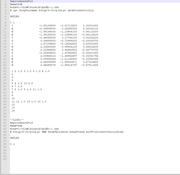
This is the input file: i have checked with Gaussian 16 User Manual and i think it's correct.
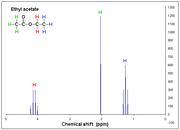
This is the experimental spectrum of ethyl acetate and the one simulated with Gaussian, for which I cannot see the spin multiplicity is shown above.
Thanks a lot
Alessio M.
Spin multiplicity is defined in Gaussian input file. It is not directly relevant to Multiwfn.
Thank you so in your opinion with MULTIWFN i wil be able to see SPIN MULTIPLICITY?
thanks a lot
Alessio Macorano
There is no evident problem. But I strongly suggesting using Multiwfn to simulate NMR spectrum, because Multiwfn is able to consider atomic equivalency of the three hydrogens in methyl group, this point is important. Also Multiwfn is also able to consider conformational weight. Please check Section 4.11.10 on how to correctly plot NMR spectum via Multiwfn, there are many examples.
Best regards,
Tian
Dear Prof Tian Lu,
yes i have used the net charge and multiplicity 0 1. here i attached two picture: one of the molecular structure and the NMR simulated spectra with Gaussian 16. level of theory: B3LYP/ 6-31+G(2d,p) 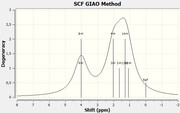
Thank you so much
Alessio M.
Dear Alessio Macorano,
Just write net charge and spin multiplicity in .gjf file according to actual situation. If ethyl acetate is not deprotonated, they should be 0 1, while if it has deprotonated, they should be -1 1.
Best,
Tian
Dear Prof Tian lu.
i'm a PhD student in medicinal chemistry at University of Milan, IT.
As a collaboration with other organic chemistry i'm trying to simulate NMR spectra with Gaussian 16, and i'm reaching you out because i'm facing with a problem on this topic.
I'd like to Ask you if you ever have any problem to see spin multiplicity in NMR spectra from Gaussian16.
Because for a simple molecule like Ethyl Acetate Gaussian doesn't show me the multiplicity spin. (The ppm are rescaleted by TMS reference and they are correct respect to sperimental spectra).
I mean it doesn't show me Doublet, triplet and so on... I have used many DFT functionals and different basis set.
I'm very courious about It. If you want i can share with you .gjf, log, chk files.
Thanks for your suggestion
Alessio Macorano