Thank you.The program is running.
Best Regards
Please check Appendix 1 of Multiwfn mauual, you need to set environmental variable so that Windows version of Gaussian can be normally run in command-line mode.
Thank you very much.
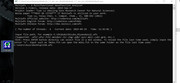
I used file C2H2.wfn in the EXAMPLE folder in the PROGRAM and I get the following error. My Gaussian 09 program runs smoothly.
No executable for file l1.exe.
Search path GAUSS_EXEDIR is ""
: No such file or directory
Gaussian running may be failed! Please manually check Gaussian input and output files in atmrad folder
Press ENTER button to continue
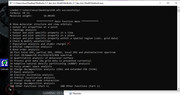
In order to calculate Hirshfeld-I charges, atomic wavefunction files must be prepared first (this can be automatically done via Multiwfn by invoking Gaussian). As prompted on screen, now the program asks you to input the calculation level (in terms of Gaussian keywords) used for calculating atomic wavefunction files. If you do not input, evidently the Hirshfeld-I calculation cannot start.
Please carefully read section 4.7.4 of the manual, in which the Hirshfeld-I charge calculation is detailedly illustrated.
Thank you reply;
I am using Multiwfn_3.7 version (downloaded from //www.umsyar.com). I am trying to calculate Fukui Functions and the Dual Descriptor.
wfn files attached.
https://1drv.ms/u/s!ApA9ZlprY_TSjQZaimQ … B?e=HpmlLW
I have stated my actions in the pictures respectively.
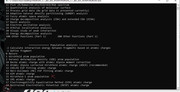
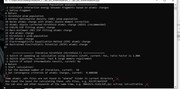
First, please make sure that you are using latest version of Multiwfn. If you are calculating Hirshfeld-I charges, note that recently I updated Multiwfn to solve a severe bug of this part of code.
If the latest version still doesn't work, please paste all of your inputted commands in your post, so that I can exactly understand how you use Multiwfn; if possible, please also upload compressed input file.
I get this error when I run the program.
''Some atomic .wfn files are not found in "atmrad" folder in current directory''
Can you help me ?please
Regards