使用CP2K过程中常用的可视化工具
使用CP2K过程中常用的可视化工具
文/Sobereva@北京科音2025-Mar-31
0 前言
使用计算化学程序过程中普遍离不开可视化问题,对如今已经非常流行、十分强大、巨快、开源免费的第一性原理程序CP2K自然也不例外。最近有人在思想家公社QQ群里问“cp2k可视化一般搭配哪个程序呢?”,针对这个问题,笔者简要罗列一下我推荐的在CP2K使用过程中的可视化程序,主要面向初学者。本文只是粗略概述,这些程序在笔者讲授的北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)中都会反复利用和详细演示具体操作。除本文说的外,还有大量其它可视化程序,比如Avogadro、gabedit、Chemcraft等,我认为对于CP2K用户来说,用好本文提到的这些就已经完全足够了。文中提到的Multiwfn可以在官网//www.umsyar.com/multiwfn下载,相关信息见《Multiwfn FAQ》(//www.umsyar.com/452)。
1 建模阶段的可视化
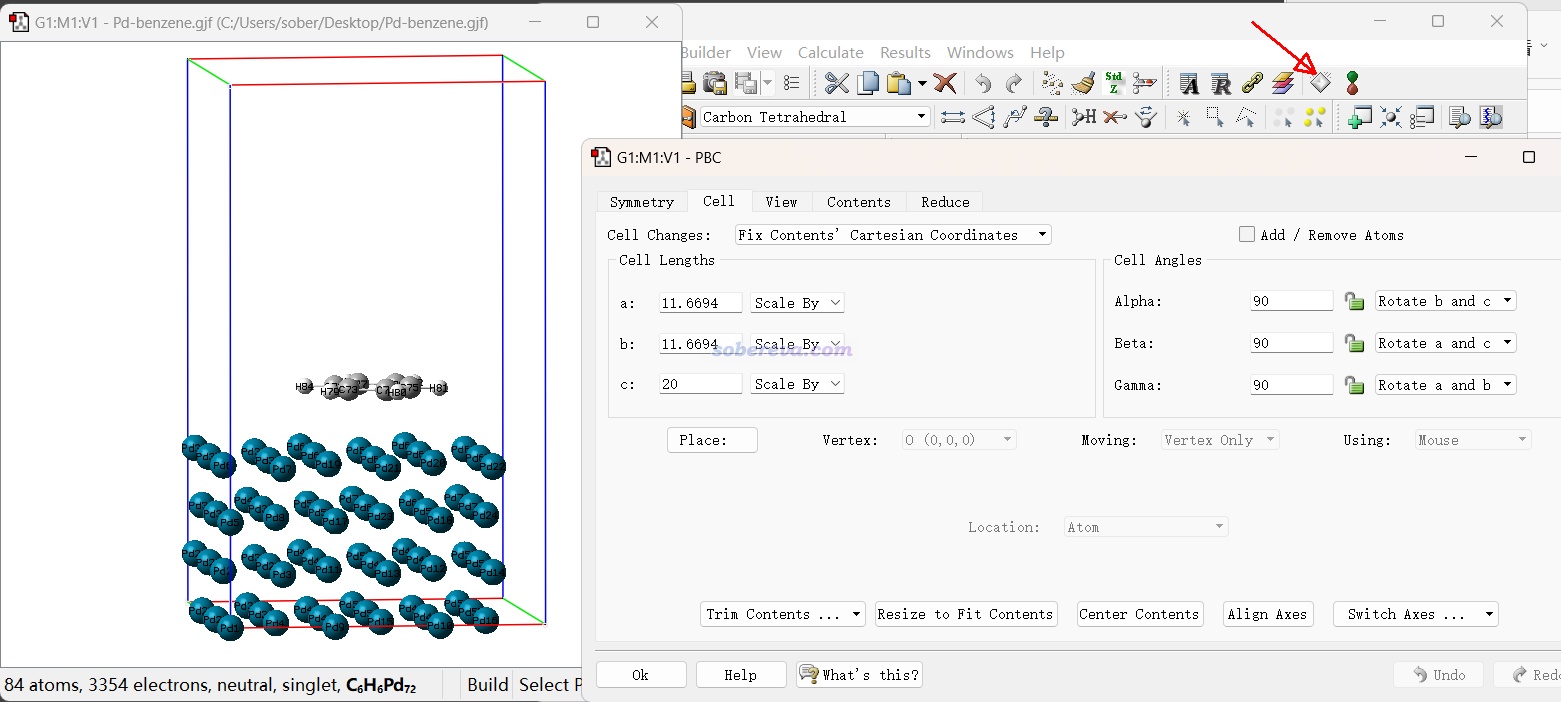
能够构建用于做第一性原理计算的周期性体系的程序很多,其中我十分推荐GaussView,这是绝大多数内行做 计算的人都会用的可视化程序,其图形界面设计得很好,所见即所得,大多数情况用这一个程序就可以很好完成CP2K计算前的建模。GaussView可以直接读取记录周期性体系原子坐标和晶胞信息的最常见的cif文件。在GaussView里构建好结构后,可以保存成Gaussian程序的输入文件(gjf),里面有原子坐标信息和晶胞的平移矢量信息(Tv开头的行),然后用Multiwfn载入这样的gjf文件后,就可以按照《使用Multiwfn非常便利地创建CP2K程序的输入文件》(//www.umsyar.com/587)文中说的创建CP2K输入文件了,整个过程相当方便。
许多人误以为GaussView只是结合Gaussian使用的,或者至多是用于 程序算孤立体系建模的,这是大错特错!由于Gaussian本来就有计算周期性的功能(但由于功能性和效率的原因,极少有人用Gaussian算周期性体系),所以Gaussian的御用图形界面GaussView也支持周期性体系,可以直接对周期性体系添加/删除/替换原子、调整键长/键角/二面角、平移/旋转片段、把其它体系粘贴到当前体系里、测量几何参数,等等,对周期性体系和对分子体系操作一样非常灵活和顺手。并且GaussView专门有个PBC editor界面,在里面可以定义晶胞参数、重新定义晶胞、修改周期性、晶轴变换、扩胞、切晶面、判断空间群、居中体系、删除晶胞外的原子、把晶胞外的原子卷入晶胞,等等,常用的操作几乎都提供了。GaussView对于周期性体系建模相当好用,然而其这方面的价值却被很多人低估,甚至有人明明不了解还胡乱贬损。
点击下图红色箭头处的按钮就可以进入前述的PBC editor界面。红、绿、蓝边框分别是晶胞的a、b、c轴。
还值得一提的是,GaussView的菜单栏的Tools中的Atom selection界面很有用。用滑框选择工具选择一批原子成为黄色后,Atom selection界面里就可以看到选中的原子序号,格式和Multiwfn程序里要求的完全一致。例如在Multiwfn里创建几何优化任务的输入文件时,若你选择要冻结某些原子,就可以把序号从这里复制出来并直接粘贴到Multiwfn的窗口中,这样实现诸如slab边缘原子的冻结设置巨方便。

Multiwfn还可以载入CP2K的输入文件、Quantum ESPRESSO的输入文件、VASP的POSCAR等格式,主功能100的子功能2里选择保存为gjf或者cif文件后,也可以用GaussView载入并观看和编辑。顺带一提,在建模方面Multiwfn也提供了很多实用功能,很多还是GaussView没有的,见《Multiwfn中非常实用的几何操作和坐标变换功能介绍》(//www.umsyar.com/610)。
2 几何优化结果的可视化
如果要看CP2K做几何优化得到的最终结构,最简单的方法是用Multiwfn载入此任务产生的restart文件,之后进入主功能0直接就看到了,例如下面的聚噻吩体系。界面上方的菜单以及图形界面右侧有一大堆选项可以调整效果,诸如视图的旋转和缩放、键的粗细、成键判断阈值、原子序号是否显示/字号/颜色、原子球大小、高亮特定原子。菜单栏的Tools列表里还有一些辅助工具,比如测量几何参数、导出所有内坐标、输出笛卡尔或分数坐标,等等。

很多时候我们还关心优化的过程,尤其是当体系结构复杂时,不观看优化轨迹的话,往往都不好判断优化过程中哪里发生了何种变化,心里没数。观看优化轨迹最好的程序是VMD,在http://www.ks.uiuc.edu/Research/vmd/可免费下载,强烈建议用VMD 1.9.3(撰此文时1.9.4还没发布正式版,其测试版的bug巨多,强烈不建议用)。CP2K的几何优化任务默认会输出.xyz文件,这是多帧的轨迹文件,里面记录了优化过程的每一帧的坐标,不熟悉xyz格式的话看《谈谈记录化学体系结构的xyz文件》(//www.umsyar.com/477)。把xyz文件载入VMD就可以观看优化轨迹了。即便任务没跑完,也可以载入此文件看看最新一步的结构。
xyz文件里不记录晶胞信息,因此在VMD里没法显示出晶胞边框,给观看周期性体系造成不便。对于不变胞的优化,可以用Multiwfn载入restart文件,此时屏幕上不仅显示晶胞信息,还会显示在VMD里定义晶胞的命令,如下图所示。

把这命令复制到VMD的命令行窗口运行,之后再输入pbc box命令,晶胞在VMD里就显示出来了,如下所示。

如果做的是变胞优化任务,晶胞在优化过程中不是固定不变的,此时建议让CP2K输出pdb格式的优化轨迹,里面记录了每一帧的晶胞信息。此文件载入VMD并显示盒子后,会看到随着轨迹的播放,盒子也实时变化。
上述说明不仅适用于优化极小点,对于dimer方法优化过渡态也是一样的。
VESTA(http://jp-minerals.org/vesta/en/)也是一个很好且免费的主要面向周期性体系的可视化工具,默认情况下的显示效果往往比VMD甚至还更好些。若想用VESTA看CP2K优化的结果,可以把restart文件载入Multiwfn,主功能100的子功能2里选择导出cif或VASP的输入文件(POSCAR),然后就能载入VESTA了。
3 反应路径的可视化
CP2K做NEB类任务需要对给定的初始结构之间插点。CP2K虽然能够自动插点(点=image),但没法在计算前进行预览。利用《sobNEB:产生CP2K的NEB的插点的方便的工具》(//www.umsyar.com/660)里介绍的笔者的sobNEB程序插点的话,可以直接产生traj.xyz轨迹文件,放到VMD里通过播放动画或者多帧叠加显示,就可以直观判断初始的NEB的image是否合理了。
NEB类任务计算过程中是好多副本一起算的,每个副本都会在计算过程中输出它所负责的那个image优化过程的xyz轨迹文件。要想观看最终收敛的NEB轨迹,需要自己把这些副本输出的轨迹的最后一帧合并在一起作为新的xyz轨迹,放到VMD里就可以观看最终的反应路径动画了。手动做比较麻烦,建议用脚本实现。北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)里专门给了个shell脚本自动做这件事情,运行后就会在当前目录下产生[项目名]_traj.xyz文件,里面记录了NEB最新一步的轨迹(即便任务还没跑完,也可以看最新的NEB轨迹是什么样),同时还会产生[项目名]_ene.txt,里面记录了最新一步的各个点的能量。下面是培训里的一个实例,对载入的[项目名]_traj.xyz文件做多帧叠加显示,直观显示了Na的迁移轨迹

4 振动分析的可视化
CP2K做完振动分析后往往需要观看振动矢量了解振动的特征。推荐做法是用《使CP2K计算的振动模式可以被GaussView观看的程序:MfakeG》(//www.umsyar.com/656)里介绍的笔者开发的MfakeG工具,把记录振动模式信息的.mol后缀的molden文件转化成仿Gaussian输出文件,之后载入GaussView就可以用Results - Vibrations选项观看振动模式了,例如下图的草酸晶体的例子

还有一种做法是使用免费的可视化程序Jmol(https://jmol.sourceforge.net),也可以载入CP2K产生的molden文件观看振动模式。不过我还是觉得GaussView用起来舒服。
如果要基于CP2K的振动分析观看振动光谱,推荐用Multiwfn。Multiwfn具有非常强大的绘制各种光谱的功能,见《使用Multiwfn绘制红外、拉曼、UV-Vis、ECD、VCD和ROA光谱图》(//www.umsyar.com/224)和《使用Multiwfn绘制NMR谱》(//www.umsyar.com/565)。Multiwfn可以载入CP2K做振动分析产生的输出文件按博文所述绘制红外光谱,如果要求计算了拉曼还可以绘制拉曼光谱。此外,Multiwfn还可以载入常规TDDFT和XAS-TDDFT任务的输出文件分别绘制UV-Vis光谱和X光吸收光谱,还可以考虑旋轨耦合效应,例如下图是CP2K+Multiwfn绘制的NaAlO2晶体的Al的K-edge XAS。Multiwfn还可以载入CP2K的NMR任务的输出文件绘制NMR谱。这些在北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)里有非常系统的讲解和丰富的例子。

5 动力学轨迹可视化
CP2K的第一性原理动力学(叫从头算动力学也行)是CP2K的杰出强项之一。和GROMACS、AMBER、NAMD等经典力场动力学程序的情况一样,VMD也是观看其动力学轨迹的不二的选择。例如培训里有个模拟质子轰击石墨烯层的例子,VMD载入轨迹后并多帧叠加显示、根据帧号着色,直观展现了模拟过程中质子的运动轨迹:

还可以自己写VMD脚本从CP2K产生的记录原子速度的xyz文件中把速度信息读入VMD,并根据速度着色,展现出质子的动能是如何传递到石墨烯板上并扩散开来的。下图黄色是打入的质子,越蓝的石墨烯原子的速度越大。

VMD不仅可以观看动力学过程中结构的变化,还可以绘制等值面图观看电子结构的变化,例如下图这个例子,直观展现了水合电子是怎么在模拟过程中出现的。

通过写VMD tcl脚本,还可以实现很复杂的分析,如下面幻灯片里讲的高温下正癸烷的裂解产物分析。PS:做动力学的,不会写分析脚本的话,稍微做深一点就会寸步难行。

北京科音分子动力学与GROMACS培训班(http://www.keinsci.com/KGMX)专门用约100页幻灯片特别完整、详细讲VMD的使用,并且还额外用近100页幻灯片深入讲VMD脚本的编写,如果想系统学习VMD的话这是极佳的途径。
笔者偶尔看到有人用OVITO程序可视化CP2K产生的轨迹,我没用过那个程序,也完全不理解为什么有人不用VMD而用OVITO,明明VMD已经可以完美地满足一切需求。笔者在答疑时看到有一些CP2K用户还被OVITO坑了:OVITO根据边缘原子位置自动确定了盒子边框,有人以为那就是实际的晶胞边界,导致对结果产生了严重误解。
6 电子结构的可视化和分析
Multiwfn可以基于CP2K的输出文件绘制效果非常理想的能带结构图,如CrO2体系:

《详谈使用CP2K产生给Multiwfn用的molden格式的波函数文件》(//www.umsyar.com/651)中介绍了怎么用CP2K产生molden文件。Multiwfn将之载入后,就可以在《使用Multiwfn绘制态密度(DOS)图考察电子结构》(//www.umsyar.com/482)讲的基础上举一反三绘制效果很好的DOS、PDOS、OPDOS、LDOS图,下图是WO3体系:

Multiwfn还能基于CP2K的molden文件观看轨道,做法可参考《使用Multiwfn观看分子轨道》(//www.umsyar.com/269)。还支持对特定k点看轨道:

Multiwfn还能对周期性体系做超级丰富的波函数分析,其中很多都是以图形方式展现的,比如可以基于《在Multiwfn中单独考察pi电子结构特征》(//www.umsyar.com/432)和《使用Multiwfn考察周期性体系的芳香性》(//www.umsyar.com/722)讲的做法计算LOL-pi格点数据,之后可以在Multiwfn里直接观看等值面;也可以导出成cube文件后,载入VMD或VESTA程序绘制,如下所示,极为生动直观地展现了一个COF体系的pi电子共轭路径

再比如下图是Multiwfn载入CP2K对硅表面计算产生的molden文件,做轨道定域化后直接显示出轨道等值面图,直观展现了体系不同位置的电子结构特征。相关信息见《Multiwfn的轨道定域化功能的使用以及与NBO、AdNDP分析的对比》(//www.umsyar.com/380)。

为避免此章太长,Multiwfn可以针对周期性体系做的巨量的可视化分析这里就不一一提及,很多分析我都写过博文,感兴趣的读者请阅读:
使用Multiwfn结合CP2K的波函数模拟周期性体系的隧道扫描显微镜(STM)图像
//www.umsyar.com/671(http://bbs.keinsci.com/thread-37740-1-1.html)
使用Multiwfn对周期性体系做键级分析和NAdO分析考察成键特征
//www.umsyar.com/719(http://bbs.keinsci.com/thread-47176-1-1.html)
使用Multiwfn结合CP2K做周期性体系的atom-in-molecules (AIM)拓扑分析
//www.umsyar.com/717(http://bbs.keinsci.com/thread-46927-1-1.html)
使用Multiwfn结合CP2K对周期性体系做电荷分解分析(CDA)
//www.umsyar.com/716(http://bbs.keinsci.com/thread-46878-1-1.html)
使用Multiwfn做IGMH分析非常清晰直观地展现化学体系中的相互作用
//www.umsyar.com/621(http://bbs.keinsci.com/thread-28147-1-1.html)
使用IRI方法图形化考察化学体系中的化学键和弱相互作用
//www.umsyar.com/598(http://bbs.keinsci.com/thread-23457-1-1.html)
使用Multiwfn结合CP2K通过NCI和IGM方法图形化考察固体和表面的弱相互作用
//www.umsyar.com/588(http://bbs.keinsci.com/thread-21742-1-1.html)
使用Multiwfn绘制分子和固体表面的距离投影图
//www.umsyar.com/589(http://bbs.keinsci.com/thread-21754-1-1.html)
使用Multiwfn图形化展现原子对色散能的贡献以及色散密度
//www.umsyar.com/705(http://bbs.keinsci.com/thread-44723-1-1.html)
使用Multiwfn做Hirshfeld surface分析直观展现分子晶体和复合物中的相互作用
//www.umsyar.com/701(http://bbs.keinsci.com/thread-43178-1-1.html)
使用CP2K结合Multiwfn对周期性体系模拟UV-Vis光谱和考察电子激发态
//www.umsyar.com/634(http://bbs.keinsci.com/thread-28006-1-1.html)
使用Multiwfn计算分子和晶体中孔洞的直径
//www.umsyar.com/643(http://bbs.keinsci.com/thread-30696-1-1.html)
使用Multiwfn计算晶体结构中自由区域的体积、图形化展现自由区域
//www.umsyar.com/617(http://bbs.keinsci.com/thread-25241-1-1.html)
使用CP2K结合Multiwfn绘制密度差图、平面平均密度差曲线和电荷位移曲线
//www.umsyar.com/638(http://bbs.keinsci.com/thread-28225-1-1.html)
此外,用VMD载入CP2K产生的Hartree势(静电势的负值),还可以实现对晶体表面静电势的可视化,一个COF体系的例子如下所示。静电势在研究静电主导的相互作用方面有重要意义,参见《静电势与平均局部离子化能相关资料合集》(http://bbs.keinsci.com/thread-219-1-1.html)里面的资料。

和静电势同等重要的是笔者提出的范德华势,适用于考察范德华作用主导的相互作用,可以由Multiwfn计算,见《谈谈范德华势以及在Multiwfn中的计算、分析和绘制》(//www.umsyar.com/551)。