帮助设定Gaussian输入文件中优化flag和MM电荷的小工具
帮助设定Gaussian输入文件中优化flag和MM电荷的小工具
A small tool to facilitate setting up optimization flags and MM charges in Gaussian input file
文/Sobereva@北京科音2016-Jun-6
A small tool to facilitate setting up optimization flags and MM charges in Gaussian input file
文/Sobereva@北京科音2016-Jun-6
近来由于计算需要写了两个小程序setopt和setcharge,一个是用来帮助设定Gaussian输入文件中的优化flag,另一个是帮助设定MM电荷。虽然程序很简单,但很有用,这里分享下。Windows版及源代码下载地址:
optflag:optflag 1.0.rar
setcharge: setcharge 1.0.rar1 optflag程序
Gaussian里optimization flag就是原子后面写0、-1来分别代表优化时允许移动和被冻结。对于ONIOM,设成同样的负值的原子还可以被当成刚性片段优化。optflag小程序可以将指定的原子的优化flag设为指定值。来看一个例子:
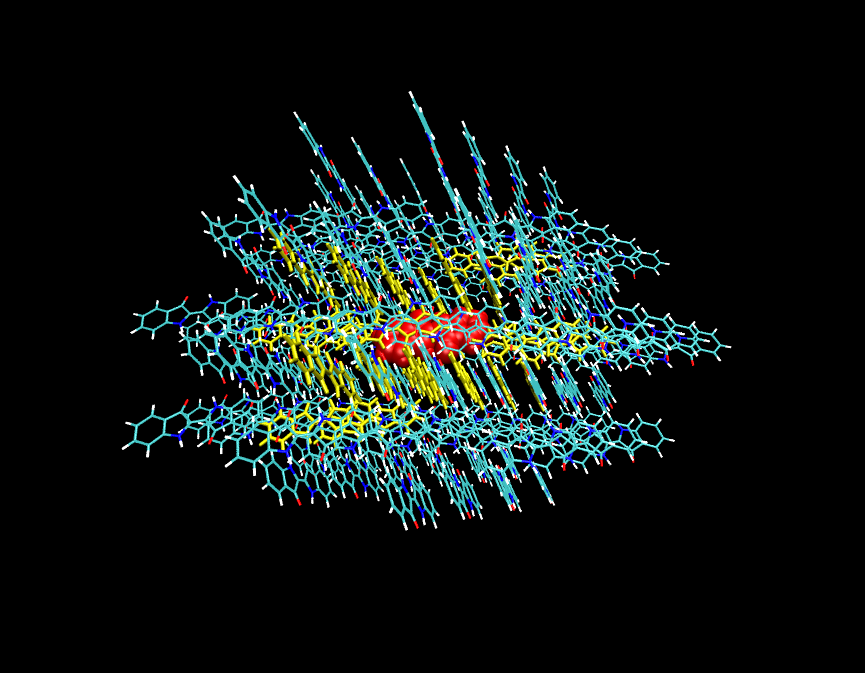
这是个靛蓝(indigo)的分子团簇,从晶体结构中截取出来的。pdb文件在此: /usr/uploads/file/20160606/20160606121956_93869.rar
我们想把中间红色的分子的周围的一圈分子,即黄色区域的靛蓝分子的优化flag设为0,允许它们在优化时调整结构,而将最外围那些以及中间的红色靛蓝分子设为冻结。
要做到这个目的,我们先用VMD生成原子序号列表。选择范围对应于:
中心分子:residue 43
中心分子+相邻部分:same fragment as within 6 of residue 43
比如要生成中心分子的序号列表,就在VMD控制台里输入atomselect top "residue 43",比如提示atomselect0,就再输入atomselect0 list得到序号列表和atomselect0 num得到原子数。然后写成索引文件center.txt,将用于作为optflag的输入,内容如下。
-30
1290 1291 1292 1293 1294 1295 1296 1297 1298 1299 1300 1301 1302 1303 1304 1305 1306 1307 1308 1309 1310 1311 1312 1313 1314 1315 1316 1317 1318 1319
第一行是原子数,之后是原子序号。由于VMD输出的序号是从0开始的,原子数要写成负值来指明这一点。以同样步骤,生成中心+相邻部分分子的索引文件small.txt。
然后用gview打开.pdb文件,保存成.gjf文件,把原子坐标那部分截出来,并且去掉(pdbname...)那一堆,保存到all.txt里面。
双击启动optflag,输入
all.txt
4530 //总原子数
0 //当前输入文件里还没有优化flag
all //选全部原子
-1 //要设成的优化flag
此时提示在当前目录产生了new.txt,打开一看,已经相对于all.txt把-1的优化flag加上了。
然后把中心+相邻部分分子优化flag设为0,启动optflag输入
new.txt
4530
1 //输入文件里已经有优化flag了
small.txt //用来选定中心分子+相邻部分的索引文件
0 //要设的优化flag
最后一次,将中心分子优化flag设为-1,启动optflag输入
new.txt
4530
1
center.txt
-1
好啦,此时得到的new.txt的内容就已经可以拷到Gaussian输入文件的坐标部分了,优化时就只有中心分子临近的一圈分子会被优化了。感兴趣的话可以把gjf文件拉进gview,进edit-atom groups,选freeze分类,然后高亮或隐藏不同的部分检查是否设定合理。
2 setcharge程序
Gaussian中用分子力学计算的时候涉及到给原子设定原子电荷,对于体系中包含许多同种分子,比如包含一大堆水分子,或者上面的例子包含一堆indigo,要给自行挨个给同种分子填上原子电荷信息很麻烦。setcharge可以帮助做这个事情。
对于上面的例子,我们要给团簇所有indigo分子设定在indigo孤立状态下算出的CHELPG电荷,并且假定中间的indigo分子带+1电荷,因此要给它赋上indigo阳离子状态下计算的CHELPG电荷。做法如下:
先对indigo在中性和+1状态下用Multiwfn计算CHELPG原子电荷(参考 //www.umsyar.com/441),然后分别写到indigo.txt和indigo+1.txt,第一行是原子数,之后是每个原子的电荷,例如indigo.txt
30
0.099919
0.519588
-0.183248
-0.016362
-0.189175
0.020601
-0.308034
0.418772
-0.659064
-0.562279
0.099919
0.519588
-0.659064
-0.183248
-0.562279
0.418772
-0.016362
-0.308034
-0.189175
0.020601
0.102237
0.100428
0.081598
0.143281
0.431738
0.102237
0.100428
0.081598
0.143281
0.431738
先给所有indigo都赋上中性下indigo的CHELPG电荷,启动setcharge,输入
new.txt //这个是上个例子最后得到的new.txt
4530
1
all //给全部原子赋上电荷
indigo.txt //含有原子电荷的文件
原先new.txt里的优化flag还都保留着。当前体系一共4530个原子,indigo.txt里有30个原子的电荷,所以会给1~30、31~60、61~90...4501~4530都依次赋上indigo.txt里的电荷。此时可以看到new.txt里的原子变成了诸如这样
C---0.308034
即曰C的原子电荷为-0.308034。前两个横杠之间是原子类型,这里没设所以留空。
然后给中心原子赋上+1态的电荷。启动setcharge,输入
new.txt //这个是上个例子最后得到的new.txt
4530
1
center.txt //中心分子的索引文件,见上例
indigo+1.txt //含有+1态CHELPG电荷的文件
然后new.txt里的信息就可以拷到gjf文件里用了。